
| Klassifikation nach ICD-10 | |
|---|---|
| Q87.0 | Angeborene Fehlbildungssyndrome mit vorwiegender Beteiligung des Gesichtes - Akrozephalosyndaktylie-Syndrome (Apert) |
| ICD-10 online (WHO-Version 2019) | |
Das Apert-Syndrom, auch Akrozephalosyndaktylie genannt, ist eine genetisch bedingte Besonderheit auf der Grundlage einer Mutation des FGFR2-Gens auf dem Chromosom 10, die zu vielfältigen körperlichen Fehlbildungen führt. Beschrieben wurde das Syndrom 1906 von dem französischen Kinderarzt Eugene Apert. Es gehört zur Gruppe der kraniofazialen Fehlbildungen, zu der auch das Carpenter-Syndrom, das Crouzon-Syndrom, das Pfeiffer-Syndrom und das Saethre-Chotzen-Syndrom gehören.
Häufigkeit
In Deutschland leben etwa 400 Personen mit dem Syndrom. Die Prävalenz beträgt etwa 1–9:100.000.
Symptome und Merkmale
möglich sind:
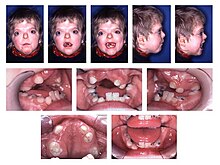

- Kopfbereich:
- Verwachsen von Schädelknochen mit der Gefahr eines Drucks auf das Gehirn und der Bildung eines Hydrocephalus
- Fehlbildung des Oberkiefers
- offene Gaumenspalte
- eingeschränktes Hörvermögen
- Sehbehinderung
- Epilepsie
- behinderte Atmung
- Hals:
- zu einem hohen Prozentsatz Segmentationsstörungen der HWS
-
Extremitäten:
- zusammengewachsene Finger und Zehen (tritt stets symmetrisch auf)
- Versteifte oder fehlende Mittelgelenke
- Knochenbau:
- Eingeschränkte Bewegung in vielen Gelenken
- Verkrümmung der Wirbelsäule (Skoliose)
- Psychologisch:
- Probleme bei sozialer und emotionaler Entwicklung ("anders sein")
Diagnostik
In der Pränataldiagnostik können typische Merkmale bereits in frühen Stadien der Schwangerschaft im Rahmen des Feinultraschalls auffallen. Die Symptome werden aber aufgrund des seltenen Auftreten des Syndroms häufig nicht richtig gedeutet. Eine Diagnose ist vorgeburtlich durch die Untersuchung des Fruchtwassers möglich, sofern ein Anfangsverdacht besteht und gezielt auf Apert-Syndrom getestet wird. Nachgeburtlich sind die meisten körperlichen Merkmale offensichtlich, eine molekulargenetische Analyse kann den Befund bestätigen.
Behandlung
Sofortige bzw. baldmöglichste Behandlung durch Spezialisten sind für die Entwicklung der Kinder und das Verhindern von Folgeschäden extrem wichtig. Zu nennen sind hier vor allem:
- Verschlossene Schädelnähte erfordern höchstwahrscheinlich Schädeloperationen, um Platzmangel für das wachsende Gehirn zu verhindern.
- Trennungsoperationen an Fingern und Zehen.
- Untersuchungen zu Atmung, Gehör, Gaumenspalte, ...
Es empfiehlt sich auch eine Kontaktaufnahme zur entsprechenden Elterninitiative.
Erbgang
Der Erbgang ist autosomal-dominant mit kompletter Penetranz und hoher Varianz (die Ausprägung gestaltet sich in Abhängigkeit von der Mutation und dem Individuum sehr variabel)
- Fibroblast growth factor receptor (FGFR2)-Gen
- verschiedene Mutationen aktivieren den Rezeptor und verstärken so vor allem das Signal zu Veränderungen in der Differenzierung von Knochen- und Knorpelgeweben
- 80 % Neumutationen, väterlicher Alterseffekt
Literatur
- Michael Eckardt: Über vier Fälle von Akrocephalosyndaktylie. Dissertation. Feder, Mediasch (Siebenbürgen) 1932, OCLC 238858702