
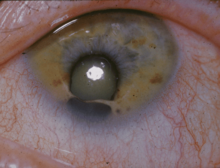
Das Katzenaugen-Syndrom (Synonym: Schmid-Fraccaro-Syndrom und Kolobom-Analatresie-Syndrom) ist eine seltene erblich bedingte Erkrankung des Menschen, deren Leitsymptome Veränderungen an den Augen (Kolobom) und eine Fehlbildung des Enddarms (Analatresie) sind.
Die Ausprägung der Symptome ist nicht vom Schweregrad der Erbgutschädigung abhängig. Die Analyse des Erbgutes weist in klassischen Fällen ein überzähliges Chromosom auf, das aus einem Fragment von Chromosom 22 besteht. Namensgebend ist die in fast allen Fällen vorkommende vertikal-ovale Spaltbildung (Kolobom) an der Regenbogenhaut (Iris), die an die Schlitzpupillen von Katzenaugen erinnert.
Epidemiologie
Das Katzenaugensyndrom ist eine angeborene Erkrankung, die durch eine Veränderung des Erbgutes bedingt ist. Der Erbgang ist autosomal-dominant. Die Häufigkeit des Katzenaugen-Syndroms liegt bei 1,35 Fällen pro 100.000 Personen. Bis 2009 wurden 105 Fälle veröffentlicht.
Ursache
Klassische Ursache dieses Syndroms ist eine Anomalie des Chromosoms 22 (Region q11.2). Charakteristisch ist bei 83 % der Fälle ein in allen Zellen des Körpers auffindbares kleines überzähliges Extrachromosom, in dem sich ein Teil der Erbinformation des Chromosoms 22 findet. Es wird angenommen, dass dieses durch eine Inversions-Duplikation entsteht, daher bizentrisch ist und an beiden Enden Satelliten trägt. Da die Erbinformation der betroffenen Region somit mehr als zweifach im Erbgut der Patienten vorliegt, spricht man von einer Tri- oder Tetrasomie.
Nicht in allen Fällen lässt sich jedoch ein Extrachromosom nachweisen. Ursächlich werden dann ein (unerkanntes) Mosaik, eine Genmutation oder eine unerkannte Translokation diskutiert.
Klinische Erscheinung

Es ist zu erkennen, dass der Dickdarm dabei blind endet, also der Anus fehlt.
Die erkennbaren körperlichen Auswirkungen (Phänotyp) der genetischen Defekte sind nicht bei allen Betroffenen gleich. Auch die für den Namen Katzenaugen-Syndrom verantwortliche angeborene vertikal-ovale Spaltbildung an der Iris liegt nicht in allen beschriebenen Fällen vor.
Als typische Symptome gelten die Analatresie und andere anorektale Fehlbildungen, ein- und beidseitig auftretende Iriskolobome sowie Präaurikularanhänge – läppchenartige Anhängsel, in der Regel aus Haut- oder Bindegewebe dicht vor der Ohrmuschel. Die Augenlidachsen der Betroffenen verlaufen häufig schräg nach außen und unten (laterokaudal). Oft sind die phänotypischen Veränderungen von einer meist geringgradigen geistigen Behinderung begleitet.
Seltener sind Fehlbildungen des Harn- und Geschlechtsapparats, wie beispielsweise doppelt angelegte Harnleiter, Wassersackniere und Verlagerung der Harnröhrenmündung.
Weiterhin kann das Skelettsystem betroffen sein, unter anderem in Form von Kleinwuchs, Fehlen des Daumens, Verformungen der Rippen, Sirenomelie und Spina bifida.
Des Weiteren gibt es eine Reihe von ebenfalls beobachteten Symptomen an den Augen, zum Beispiel großer Augenabstand, Schielen, Mongolenfalte, Grauer Star, retinale Dysplasie und krankhafte Verkleinerung des Augapfels.
Auch Herzfehler wie die Fallot-Tetralogie oder isolierte Septumdefekte von Vorhof und Kammer sowie die Beteiligung weiterer Organe wie Gehirn (Oligophrenie), Gallengangssystem (Atresie der Gallenblase) und Dickdarm (Morbus Hirschsprung) sind beschrieben.
Diagnose und Differentialdiagnose
| Klassifikation nach ICD-10 | |
|---|---|
| Q92.8 | Sonstige näher bezeichnete Trisomien und partielle Trisomien der Autosomen |
| Q13.0 | Angeborene Fehlbildungen des vorderen Augenabschnittes – Iriskolobom |
| Q42.- | Angeborene(s) Fehlen, Atresie und Stenose des Dickdarmes |
| ICD-10 online (WHO-Version 2019) | |
Diagnose
Bei Vorliegen der typischen Leitsymptome (Kolobom und Analatresie) kann die Diagnose Katzenaugen-Syndrom klinisch – also ohne Analyse des Erbgutes – gestellt werden.
Die Diagnose von Art und Umfang des im Einzelfall genetischen Defektes erfolgt mittels Chromosomenanalyse. Das verlässlichste Kriterium für die Diagnose stellt das Vorhandensein des Extrachromosoms dar.
Wird in einem Fall eine Genanalyse durchgeführt und eine entsprechende Tri- oder Tetrasomie 22 festgestellt, dann wird der Begriff jedoch auch beim Fehlen der typischen Leitsymptome verwendet, man spricht dann aber von einem „inkompletten“ Katzenaugen-Syndrom. Umgekehrt kann bei Patienten mit typischen klinischen Zeichen, aber ohne nachweisliche Erbgutveränderungen, diese Diagnose gestellt werden, insbesondere, wenn in solchen Fällen auffällig lange Daumen und eine von der Norm abweichende Stellung von Händen und Füßen vorliegen.
Differentialdiagnose
Angeborene Störungen, die meist mit einer Minderbegabung und Missbildungen einhergehen, durch Mutationen größerer Genomabschnitte verursacht werden und so zu einem Dosis-Effekt zahlreicher Gene führen, nennt man auch Genomische Erkrankungen. Mutationen, die mehrere Megabasenpaare umfassen, können nicht selten zytogenetisch charakterisiert werden, da durch ein solches Ereignis ganze Chromosomenbanden verändert werden. Solche Störungen sind in ihrer Summe keine seltenen Ereignisse. Sie machen bis zu etwa 1 % aller kongenitalen Störungen aus. Die häufigste Störung dieser Art ist das DiGeorge-Syndrom (Syn.: Velocardiofaziales Syndrom), das ähnlich wie das Katzenaugen-Syndrom durch eine Mutation im Bereich der Chromosomenbande 22q11 verursacht wird.
Das DiGeorge-Syndrom und das Conotruncal anomaly face syndrome stellen zusammen mit dem sehr seltenen der(22)-Syndrom sowie dem Katzenaugen-Syndrom einen Störungscluster dar, der mit dem Begriff „Genomische Störungen im Bereich von 22q11“ beschrieben wird. Diese drei kongenitalen Erkrankungen sind diejenigen, die am häufigsten mit Veränderungen im Bereich von Chromosom 22q11 in Verbindung gebracht werden.
Indirekt werden auch die Schizophrenie und Bipolare Störungen mit Veränderungen in 22q11 in Verbindung gebracht, da sich dort das Gen für das Enzym Catechol-O-Methyltransferase befindet und Patienten mit einer 22q11-Deletion und einem DiGeorge-Syndrom in etwa 30 % der Fälle eine Psychose entwickeln. Eine ähnliche Situation wie im Falle des DiGeorge-Syndroms (angeborene Störung mit Haploinsuffizienz und Psychose) findet sich auch beim Williams-Beuren-Syndrom. Ein Syndrom der Mikroduplikationen und Mikrotriplikationen einer 22q11.2-Region scheint ein eigenständiges Syndrom zu sein. Weitere mit dieser Region zusammenhängende Störungen sind das autosomal-dominante „Giant platelet Syndrom“, das Fechtner-Syndrom und das Rhabdoid-Prädispositions-Syndrom.
Aufgrund der Vielfalt der beschriebenen Störungen sei an dieser Stelle nur der vermutliche Mechanismus von Katzenaugen-, DiGeorge- und der(22)-Syndrom kurz beschrieben. Bei allen drei Erkrankungen liegt eine Fehlverteilung der Region 22q11 vor. Beim DiGeorge-Syndrom gibt es eine sogenannte Haploinsuffizienz der betroffenen Gene. Von der Region 22q11 existiert nur ein gesundes Allel. Genauer gesagt liegt hier eine Deletion des dem Telomer zugewandten Abschnittes von 22q11 vor. Beim der(22)-Syndrom gibt es bei den betroffenen Individuen eine zusätzliche Kopie der dem Zentromer zugewandten Region von 22q11. Beim Katzenaugen-Syndrom findet man regelmäßig zwei zusätzliche Kopien des zentromeren Abschnittes von 22q11. Diese zusätzlichen Kopien liegen üblicherweise in Form eines zusätzlichen bizentrischen Chromosoms vor. Dabei findet man drei verschiedene Varianten dieses Zusatzchromosoms. Eine Analyse der in dieser Region liegenden Gene ergab Hinweise darauf, dass der Dosis-Effekt beim DiGeorge-Syndrom etwa 24 und beim Katzenaugen-Syndrom etwa 14 Gene betrifft.
Prognose und Therapie
Die Prognose des Katzenaugen-Syndroms ist maßgeblich von Schweregrad und Art der Fehlbildungen an Herz und Nieren sowie dem Ausmaß der geistigen Entwicklungsstörung abhängig. Bei Patienten mit nur wenigen oder leichten Symptomen ist die Lebenserwartung nicht eingeschränkt.
Vorbeugend wird eine Familienberatung nach Chromosomenanalyse empfohlen. Hierbei sind Fälle mit dem Nachweis eines Extrachromosoms von solchen ohne Nachweis zu unterscheiden. Die Geschwister von ersteren gelten, sofern bei ihnen kein Extrachromosom vorhanden und keine weiteren Fälle (auch bei entfernteren) Blutsverwandten bekannt sind, als nicht gefährdet, die Erkrankung weiterzugeben. In allen anderen Fällen besteht ein potenziell erhöhtes Risiko dafür.
Im zentralen Interesse der Therapie stehen, sofern vorhanden, Analatresie sowie Fehlbildungen des Herzens und der Nieren. Die Analatresie wird chirurgisch korrigiert, da sie zu einem raschen Tod des Neugeborenen führen würde. Die Behandlung der Fehlbildungen an Herz und Nieren ist abhängig von deren Art. Nicht korrigierbare Fehler können die Lebenserwartung erheblich einschränken.
Geschichte und Herkunft des Namens
Der Name Katzenaugen-Syndrom bezieht sich auf die für dieses Erkrankungsmuster besonders typische und in fast allen Fällen vorliegende angeborene vertikal-ovale Spaltbildungen an der Iris, die den Augen der Betroffenen ein „katzenartiges“ Aussehen verleiht.
Ein eigenständiges Syndrom aus Iriskolobom und Analatresie wurde bereits 1878 durch Otto Haab erstmals beschrieben (daher auch der Name Kolobom-Analatresie-Syndrom), aber erst 1969 als eigenständige Erkrankung abgegrenzt. Zu diesem Zeitpunkt war das mögliche Vorliegen eines Extrachromosoms jedoch bereits bekannt, die Erstbeschreibung eines bei einem Patienten mit Kolobom und Analatresie auftretenden Extrachromosomes war 1965 publiziert worden.Werner Schmid (Zürich) und Marco Fraccaro (Pavia) waren maßgeblich an dieser Entdeckung beteiligt (daher auch die Bezeichnung Schmid-Fraccaro-Syndrom). 1972 wurde von Erica Bühler und Mitarbeitern erstmals erkannt, dass das Extrachromosom vom Chromosom 22 abstammt und es sich somit um eine zumindest partielle Trisomie 22 handelt.
Literatur
- J. Kunze: Atlas der klinischen Syndrome: Für Klinik und Praxis. Schattauer Verlag, 2009, ISBN 978-3-7945-2657-4, S. 94–95.
- H. Chen: Atlas of genetic diagnosis and counseling. Humana Press, 2006, ISBN 1-58829-681-4, S. 136–138.
- A. J. Augustin: Augenheilkunde. Springer, 2007, ISBN 978-3-540-30454-8, S. 493.
Weblinks
- Katzenaugen-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)