
| Klassifikation nach ICD-10 | |
|---|---|
| C92.0 | Akute myeloische Leukämie |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation nach ICD-O-3 | |
|---|---|
| 9861/3 | Akute myeloische Leukämie o.n.A. |
| ICD-O-3 erste Revision online | |
Die akute myeloische Leukämie (AML) ist eine maligne (bösartige) Erkrankung des blutbildenden Systems, und zwar der Myelopoese, also des Teils des blutbildenden Systems, der für die Bildung von Granulozyten, Monozyten, Erythrozyten und Megakaryozyten verantwortlich ist. Sie führt zu einer zum Teil massiven Vermehrung unreifer Vorstufen der Myelopoese im Knochenmark und in der Mehrzahl der Fälle auch im Blut (Leukozytose).
Geschichte
Der Begriff „Leukämie“ wurde 1845 von Rudolf Virchow geprägt, der damals das Krankheitsbild einer chronischen myeloischen Leukämie beschrieb. Die Entwicklung von Färbeverfahren für Blutausstriche im Jahre 1891 durch Paul Ehrlich führte zu neuen Erkenntnissen über die Morphologie der akuten Leukämien und ermöglichte in der Folge die Abgrenzung der myeloischen von der lymphatischen Leukämie (Naegeli 1900).
Epidemiologie
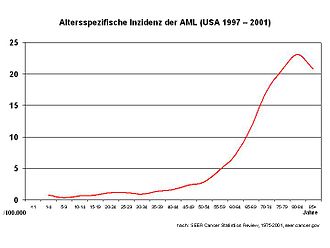
Die AML ist eine seltene Erkrankung mit einer Inzidenz von etwa drei Neuerkrankungen/100.000 im Jahr. In Deutschland treten etwa 3.600 Neuerkrankungen pro Jahr auf. Sie ist überwiegend eine Erkrankung des höheren Lebensalters, das mediane Alter bei Diagnosestellung liegt bei 63 Jahren. Die AML macht etwa 80 % aller akuten Leukämien bei Erwachsenen aus. Männer sind etwas häufiger betroffen als Frauen (Verhältnis 1,4:1). Im Kindesalter haben nur 15 bis 20 % der Patienten mit einer akuten Leukämie eine AML. Allerdings handelt es sich bei der seltenen akuten Leukämie im Neugeborenenalter meist um eine AML.
Ursachen und Entstehung
Erworbene AML
Bekannte Risikofaktoren für die Entwicklung einer AML sind eine Exposition gegenüber einer hohen Dosis ionisierender Strahlung (z. B. nach den Atombombenexplosionen in Hiroshima und Nagasaki) und eine langjährige chronische Belastung mit Benzol. Auch nach Anwendung bestimmter Zytostatika, wie Alkylanzien und Etoposid, kann es nach einer Latenzzeit von mehreren Jahren zur Entwicklung einer AML kommen. Auch das Rauchen spielt eine Rolle bei der Entstehung der AML. In vielen Fällen bleibt die Ursache jedoch unklar.
Genetische Faktoren resp. Ursachen
Die Mechanismen, die zur Entwicklung einer AML führen, sind Gegenstand der aktuellen Forschung. Man geht heute davon aus, dass am Anfang der Leukämieentstehung genetische Veränderungen in einer einzelnen hämatopoetischen Vorläuferzelle stehen. Diese Veränderungen führen zu einer klonalen Vermehrung unreifer Zellen, die die Fähigkeit zur Ausreifung verloren haben. Bei zahlreichen bei der AML bekannten chromosomalen Aberrationen (siehe unten: Abschnitt Zytogenetik) sind häufig Gene involviert, die in der normalen Zellregulation eine wichtige Rolle spielen. Durch Translokationen entstehen zum Teil neue Fusionsgene, die an der Leukämieentstehung beteiligt sind, z. B. PML/RARα bei t(15;17), AML1/ETO bei t(8;21).
Eine AML tritt gehäuft bei einigen genetischen Erkrankungen wie z. B. dem Down-Syndrom oder Fanconi-Anämie auf.
Klinik und Verlauf
Die leukämischen Zellen breiten sich in Knochenmark und Blut aus und können auch Lymphknoten, Milz sowie weitere Organe, in seltenen Fällen auch das Zentralnervensystem infiltrieren. Unmittelbare Folge ist eine Verdrängung der normalen Blutbildung (Hämatopoese). Es entsteht ein Mangel an Erythrozyten (Anämie), Blutplättchen (Thrombozytopenie) und funktionsfähigen reifen Granulozyten (Lymphopenie).
Die Symptome bei AML sind vorwiegend auf die Knochenmarkinsuffizienz zurückzuführen. Es handelt sich häufig um ein akutes Krankheitsbild. Die entsprechenden Symptome entwickeln sich häufig innerhalb weniger Tage. Zu den Symptomen zählen:
-
B-Symptomatik
- Fieber
- Nachtschweiß
- Gewichtsverlust (mind. 10 % innerhalb der letzten 6 Monate)
- Symptome einer Blutarmut (Anämie)
- Allgemeine Schwäche, Krankheitsgefühl, Blässe
- zunehmende Gerinnungsstörungen in Folge der Thrombozytopenie (Mangel an Blutplättchen)
- Petechien, Nasenbluten (Epistaxis), Hämatomen oder Schleimhaut- oder Zahnfleischblutungen
- Infektneigung durch einen Mangel an funktionsfähigen Immunzellen (Lymphopenie)
- Infektionen unterschiedlicher Lokalisation wie z. B. Pneumonie („Lungenentzündung“), Tonsillitis („Mandel-Entzündung“) oder unklares Fieber
- Entzündung der Mundschleimhaut, Mundsoor
- Symptome einer leukämischen Organinfiltration
- Meningeosis leucaemica: Die unreifen Blasten lagern sich in den Hirnhäuten ab. Folge können Meningitis-ähnliche Symptome sein. Dieser Verlauf ist bei der AML insgesamt selten und tritt wesentlich häufiger bei der ALL auf.
- Hautinfiltrationen (sog. Chlorom)
- Spleno- und Hepatomegalie: Durch die Verdrängung der physiologischen Hämatopoese (Blutbildung) im Knochenmark kann es zur extramedullären Blutbildung, d. h. eine Blutbildung außerhalb des Knochenmarks, kommen. Dies geschieht bevorzugt in der Milz und der Leber. Darüber hinaus kann es zur Organinfiltration durch leukämische Blasten kommen. Organvergrößerungen (und zum Teil Schmerzen) können die Folge sein.
- Infiltrationen der Haut- und Mundschleimhaut (Gingivahyperplasie), v. a. bei der M4/5-Variante (s. u.)
- Lymphknotenschwellungen (häufiger bei der ALL)
- Komplikationen einer AML
- Leukostase-Syndrom: Durch eine übermäßige Zahl unreifer Leukozyten im Blut kann es zu Mikrozirkulationsstörungen kommen, d. h. die Blutversorgung durch die Kapillaren ist stark eingeschränkt. Betroffene Organe sind vor allem die Lunge und das zentrale Nervensystem. Entsprechende Symptome sind u. a. Atemnot, Sauerstoffunterversorgung (Hypoxie) und Tachypnoe sowie Kopfschmerzen, Schwindel und Bewusstseinsstörungen.
- Tumorlyse-Syndrom: Durch einen raschen Zerfall von Tumorzellen (z. B. in Folge einer Chemotherapie) kommt es zur Freisetzung intrazellulärer Metabolite, wie Harnsäure, Phosphat und Kalium. Es resultieren eine Hyperurikämie, Hypocalciämie (durch Bindung des Calcium an das überschüssige Phosphat) und Hyperkaliämie. Nierenschäden und Herzrhythmusstörungen können die Folge sein.
Häufig auftretende weitere Befunde sind:
- In den meisten Fällen Leukozytose, manchmal auch normale oder sogar erniedrigte Leukozytenzahl
- Eine Leukozytose sollte nicht mit einer Lymphozytose verwechselt werden. Die Lymphozyten gehören zur Gruppe der Leukozyten (weiße Blutkörperchen). Die Lymphozyten werden durch die übermäßige Proliferation (Zellvermehrung) der entarteten myeloischen Zellen in ihrer Bildung verdrängt, sodass trotz einer Leukozytose (Vermehrung weißer Blutkörperchen) eine Lymphopenie (Mangel an Lymphozyten) bestehen kann.
- Auftreten von Blasten im Differentialblutbild
- Erhöhung von LDH, BSG und Harnsäure
- plasmatische Gerinnungsstörungen
- Während es durch einen Mangel an Thrombozyten bei der AML regelhaft zu Störungen der primären (zellulären) Blutgerinnung kommt, kann es – vorwiegend bei Promyelozyten-Leukämie (M3 der AML, s. u.) – ferner zu einer Störung der plasmatischen Gerinnung kommen
Unbehandelt schreitet die Erkrankung schnell voran und führt nach einigen Wochen zum Tode, meist aufgrund von unbeherrschbaren Infektionen oder Blutungen.
Diagnostik und Klassifikation
Die Verdachtsdiagnose einer akuten Leukämie ergibt sich aus den klinischen Symptomen und dem Blutbild einschließlich Differentialblutbild. Die Diagnose wird gesichert durch eine Untersuchung des Knochenmarks (siehe Knochenmarkpunktion). Entscheidend für die Behandlung und Prognose ist die Abgrenzung einer AML von einer akuten lymphatischen Leukämie (ALL).
Die moderne Diagnostik beruht auf der Kombination morphologischer und zytochemischer Befunde, ergänzt durch die Immunphänotypisierung und Zytogenetik und gegebenenfalls molekulare Diagnostik. In Zukunft wird vermutlich die Analyse von Genexpressionsprofilen mittels der Microarray-Technik zunehmende Bedeutung erlangen.
Die Diagnose einer AML erfordert:
- den Nachweis eines Anteils unreifer Blasten von mindestens 30 % (FAB-Klassifikation – French-American-British) bzw. 20 % (WHO-Klassifikation) im Knochenmark.
- die Zuordnung der Blasten zur myeloischen Reihe durch zytochemische Untersuchung und/oder Immunphänotyp.
- die weitere Zuordnung zu einem AML-Subtyp entsprechend der FAB-Klassifikation oder WHO-Klassifikation.
Morphologie und Zytochemie
Grundlage der Diagnostik ist die mikroskopische Untersuchung von Knochenmarkausstrichen. Charakteristische Merkmale wie z. B. der Nachweis von Auerstäbchen ermöglichen die Zuordnung der Blasten zur myeloischen Reihe. Bei Auerstäbchen handelt es sich um feine, stäbchenförmige Granula oder große, ovale bis elliptiforme Einschlüsse (Auer-Körper) im Zytoplasma unreifer Leukämiezellen. Gegebenenfalls mit Hilfe zusätzlicher zytochemischer Untersuchungen (Peroxidase, Esterase, PAS-Reaktion) gelingt in der Mehrzahl der Fälle die Abgrenzung einer AML von einer ALL und die Einordnung entsprechend der FAB-Klassifikation.

| FAB-Subtyp | Bezeichnung | Zytogenetische Aberrationen |
|---|---|---|
| M0 | Akute myeloische Leukämie mit minimaler Differenzierung | – |
| M1 | Akute myeloische Leukämie ohne Ausreifung | – |
| M2 | Akute myeloische Leukämie mit Ausreifung | t(8;21) |
| M3 | Akute Promyelozyten-Leukämie (APL) | t(15;17) |
| M3v | Akute Promyelozyten-Leukämie, mikrogranuläre Form | t(15;17) |
| M4 | Akute myelomonozytäre Leukämie | – |
| M4Eo | Akute myelomonozytäre Leukämie mit Eosinophilie | inv(16) |
| M5a | Akute Monoblasten-Leukämie | – |
| M5b | Akute Monozyten-Leukämie | – |
| M6 | Akute Erythroleukämie | – |
| M7 | Akute Megakaryoblasten-Leukämie | – |
Eine Weiterentwicklung der FAB-Klassifikation stellt die WHO-Klassifikation dar, die häufige chromosomale Aberrationen bei der AML mit einbezieht. Die Promyelozytenleukämie (FAB M3 bzw. M3v) weist klinische, biologische und auch therapeutische Besonderheiten auf und wird in einem eigenen Artikel besprochen.
Immunphänotypisierung
Die Immunphänotypisierung hat bei der AML vorwiegend bestätigenden Charakter, kann aber in Zweifelsfällen wichtige zusätzliche Informationen liefern. Mit Hilfe von markierten monoklonalen Antikörpern wird die Expression membranständiger Oberflächenmoleküle auf den Leukämiezellen untersucht. In den meisten Fällen handelt es sich um Differenzierungs-Antigene, die auch im Verlauf der normalen Hämatopoese exprimiert werden und in der sogenannten CD-Nomenklatur zusammengefasst werden. Die für die Charakterisierung einer AML wichtigsten Antigene sind in folgender Tabelle zusammengefasst.
| Antigen | |
|---|---|
| Pan-myeloisch | CD13, CD33, CD65s |
| Granulozytär, monozytär | CD15, CD14, CD64 |
| Vorläufer-Zellen | CD34, CD117, CD7 |
Zytogenetik
Bei den meisten Patienten mit AML lassen sich erworbene numerische und strukturelle chromosomale Aberrationen in den Leukämiezellen nachweisen. Einige dieser Aberrationen sind eng mit einem bestimmten morphologischen und klinischen Subtyp verbunden bzw. definieren diesen. In den letzten Jahren ist die diagnostische, prognostische und therapeutische Bedeutung der zytogenetischen Veränderungen bei AML zunehmend klar geworden. Häufige chromosomale Aberrationen sind:
| Aberration | Besondere Merkmale |
|---|---|
| +8 | Häufigste numerische Chromosomenaberration bei AML |
| t(8;21) | Häufigste strukturelle Chromosomenaberration bei AML |
| t(15;17) | Spezifisch für Promyelozytenleukämie |
| t(9;22) | sog. „Philadelphia-Chromosom“ |
| inv(16) | M4Eo |
Therapie und Prognose
Prognostische Faktoren
Man unterscheidet eine de-novo-AML von einer sekundären AML. Beide Formen unterscheiden sich hinsichtlich der Biologie der Erkrankung und dem therapeutischen Ansprechen. Von einer sekundären AML spricht man, wenn der Erkrankung eine hämatologische Erkrankung, z. B. ein myelodysplastisches Syndrom (MDS), vorangegangen ist oder eine Exposition gegenüber Zytostatika oder Strahlen bestand. Die sekundäre AML weist einige prognostisch ungünstige zytogenetische Veränderungen auf und spricht auf die Therapie schlechter an.
Anhand einer Reihe von prognostischen Faktoren lässt sich das Ansprechen auf eine Therapie abschätzen. Diese Faktoren sind allerdings nicht voneinander unabhängig, z. B. sind komplexe zytogenetische Aberrationen bei sekundärer AML häufiger.
Prognostisch ungünstig sind u. a.:
- Hohes Alter und/oder schlechter Allgemeinzustand bei Diagnosestellung
- Hohe Leukozytenzahl
- Vorangegangene hämatologische Erkrankung (z. B. MDS, Blastenschub bei CML), sekundäre AML
- FAB M0, FAB M6, FAB M7
- Ungünstige Zytogenetik (Trisomie 8, Aberrationen von Chromosom 5 oder 7, t(6;9), t(9;22), 11q23-Aberrationen, komplexe zytogenetische Aberrationen)
- Sekundäre Leukämie
Prognostisch günstige Faktoren sind:
- Promyelozyten-Leukämie (FAB M3)
- Günstige Zytogenetik: t(15;17), inv(16), t(8;21), normaler Karyotyp
Therapiedurchführung
Grundlage der Therapie der AML ist eine intensive Chemotherapie. Ziel der sog. Induktionstherapie ist das Erreichen einer kompletten Remission, d. h. einer Beseitigung aller Krankheitssymptome mit Normalisierung des Blutbildes und Beseitigung der pathologischen Zellpopulation im Knochenmark (Blasten < 5 %). Die Behandlung besteht aus mehrtägigen Therapieblöcken, die mehrfach (zwei bis drei Mal) wiederholt werden. Bei diesen kommt der Patient jeweils in sogenannte „Isolation“; dabei ist die Zahl der Leukozyten so niedrig, dass jeder Infekt unter Umständen tödlich sein kann, was absolute Mundschutzpflicht beim Patientenumgang bedingt. Wichtige Medikamente sind Cytarabin und die Anthracycline Daunorubicin bzw. Idarubicin. Gegebenenfalls wird zusätzlich Thioguanin gegeben. Ist eine komplette Remission erreicht (meist nach 1–2 Therapieblöcken), folgt die Postremissionstherapie. Sie umfasst die Konsolidierungstherapie (hochdosiertes Cytarabin) und bei der Akuten Promyelozytenleukämie eine Erhaltungstherapie (All-trans Retinol, 6-Mercaptopurin und Methotrexat). Als weitere Therapieverfahren kommen die allogene oder autologe Stammzelltransplantation in Betracht.
Azacitidin ist zugelassen für die Therapie einer akuten myeloischen Leukämie mit 20 – 30 % Blasten und Mehrlinien-Dysplasie gemäß Klassifikation der WHO.
Der Stellenwert neuer Therapieansätze, wie beispielsweise das Immuntoxin Gemtuzumab-Ozogamicin, ist bisher noch unklar.
In der Begleittherapie spielt der Ersatz von Blutbestandteilen (Erythrozyten, Thrombozyten), evtl. der Einsatz von Wachstumsfaktoren für Granulozyten (z. B. G-CSF) und die Bekämpfung von Infektionen mittels Antibiotika gegen bakterielle Infektionen und Antimykotika gegen Pilzinfektionen eine wesentliche Rolle.
Die Chemotherapie kann zusätzlich durch die Gabe von all-trans Retinoinsäure (ATRA) unterstützt werden. Die Evidenz ist sehr ungewiss bezüglich der Wirkung von all-trans Retinoinsäure zusätzlich zu einer Chemotherapie bei Patienten mit einer akuten lymphatischen Leukämie auf Durchfall, Übelkeit/Erbrechen und das Herz betreffende Toxizität dritten/vierten Grades. Außerdem führt all-trans Retinoinsäure als Ergänzung zu einer Chemotherapie lediglich zu einer kleinen oder keiner Veränderung bezüglich der Mortalität, der Rückfälle, dem Fortschritt der Erkrankung, der Mortalität während der Studie und Infektionen dritten und vierten Grades.
Bei der Chemotherapie und der Stammzelltransplantation ergeben sich Nebenwirkungen wie Blutungen oder Transplantat-gegen-Wirt Reaktionen (nur bei Stammzelltransplantation). Cochrane Haematology hat daher einige Übersichtsarbeiten erstellt, um zu evaluieren, wie diese Nebenwirkungen therapiert oder vermieden werden können. Estcourt et al. haben in den Jahren 2012 und 2015 Cochrane-Übersichtsarbeiten mit randomisierten kontrollierten Studien erstellt, um herauszufinden, welche Nutzung von Thrombozytentransfusionen die wirksamste ist, um Blutungen bei Patienten mit hämatologischen Erkrankungen zu verhindern, wenn sie eine Chemotherapie oder eine Stammzelltransplantation erhalten. Es zeigten sich dabei verschiedene Ergebnisse beispielsweise bezüglich der Anzahl der Blutungsereignisse. Es wurden Cochrane-Übersichtsarbeiten mit randomisierten kontrollierten Studien erstellt, um die Sicherheit und Wirksamkeit von mesenchymalen Stromazellen (MSC) bei Patienten mit einer Graft-versus-Host-Reaktion (GvHD) zu messen. Die Evidenz ist sehr ungewiss bezüglich der Wirkung von mesenchymalen Stromazellen für die Behandlung von Graft-versus-Host Erkrankungen nach Stammzelltransplantationen auf die Vollremission (= komplettes Verschwinden) von akuten und chronischen Graft-versus-Host Reaktionen bei therapeutischer Anwendung. Mesenchymale Stromazellen verursachen eventuell nur eine geringe oder keine Veränderung bezüglich der Gesamtmortalität, der Rückkehr der malignen Erkrankung und der Inzidenz der akuten und chronischen Graft-versus-Host Reaktion bei prophylaktischen Zwecken.
Therapieergebnisse
Mit der Induktionstherapie gelingt es, je nach Patientenauswahl, bei etwa 70 % der Patienten mit AML eine komplette Remission zu erreichen.
Die Therapieergebnisse bei älteren Patienten (> 60 Jahre) sind deutlich schlechter, die Rate kompletter Remissionen liegt hier zwischen 30 % und 60 %. Ursache sind nicht nur Begleiterkrankungen im höheren Lebensalter, die zu Komplikationen führen, sondern auch das vermehrte Auftreten prognostisch ungünstiger Faktoren wie ungünstige Zytogenetik oder Sekundärleukämien nach vorangegangenem Myelodysplastischen Syndrom. Leider ist bei der Mehrheit der Patienten mit einem Rückfall zu rechnen, etwa 15 %–25 % erreichen Langzeitremissionen nach konventioneller Chemotherapie und können als geheilt angesehen werden. Günstiger sind die Ergebnisse nach allogener Stammzelltransplantation. In einer bundesweiten klinischen Studie mit über 220 Patienten, die in den Jahren zwischen 2003 und 2009 durchgeführt wurde, wurde der Einsatz von niedrig dosiertem Decitabin für die Therapie von älteren Patienten untersucht.
Wesentlich bessere Therapieresultate ergeben sich für die Promyelozytenleukämie.
Supportivtherapie
Die Therapie der Erkrankung kann durch supportive Maßnahmen unterstützt werden. Dies kann unter anderem die körperliche Betätigung sein. Die Evidenz ist sehr ungewiss bezüglich des Effekts von körperlicher Betätigung auf Angst und schwere unerwünschte Ereignisse. Körperliche Betätigung verursacht eventuell nur eine geringe oder keine Veränderung bezüglich der Mortalität, der Lebensqualität und der körperlichen Funktion. Körperliche Betätigung verursacht eventuell eine schwache Verringerung von Depressionen.
Literatur
- Übersichtsartikel
- J. E. Rubnitz, H. Inaba: Childhood acute myeloid leukaemia. In: British Journal of Haematology. Band 159, Nummer 3, November 2012, S. 259–276, ISSN 1365-2141. doi:10.1111/bjh.12040. PMID 22966788. PMC 3468705 (freier Volltext). (Review).
- U. Creutzig, M. M. van den Heuvel-Eibrink, B. Gibson, M. N. Dworzak, S. Adachi, E. de Bont, J. Harbott, H. Hasle, D. Johnston, A. Kinoshita, T. Lehrnbecher, G. Leverger, E. Mejstrikova, S. Meshinchi, A. Pession, S. C. Raimondi, L. Sung, J. Stary, C. M. Zwaan, G. J. Kaspers, D. Reinhardt: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. In: Blood. Band 120, Nummer 16, Oktober 2012, S. 3187–3205, ISSN 1528-0020. doi:10.1182/blood-2012-03-362608. PMID 22879540. (Review).
- B. Löwenberg, J. R. Downing, A. Burnett: Acute myeloid leukemia. In: The New England journal of medicine. Band 341, Nummer 14, September 1999, S. 1051–1062, ISSN 0028-4793. doi:10.1056/NEJM199909303411407. PMID 10502596. (Review).
- Leitlinien
- S1-Leitlinie Akute myeloische Leukämie (AML) im Kindesalter der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH). In: AWMF online (Stand 2013)
- Sonstiges
- J. M. Bennett, D. Catovsky, M. T. Daniel, G. Flandrin, D. A. Galton, H. R. Gralnick, C. Sultan: Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. In: Annals of internal medicine. Band 103, Nummer 4, Oktober 1985, S. 620–625, ISSN 0003-4819. PMID 3862359.
- J. W. Vardiman, N. L. Harris, R. D. Brunning: The World Health Organization (WHO) classification of the myeloid neoplasms. In: Blood. Band 100, Nummer 7, Oktober 2002, S. 2292–2302, ISSN 0006-4971. doi:10.1182/blood-2002-04-1199. PMID 12239137. (Review).
Weblinks
- Kompetenznetz Pädiatrische Onkologie und Hämatologie (KPOH)/kinderkrebsinfo.de: Akute myeloische Leukämie (AML) doi:10.1591/poh.patinfo.aml.1.20060414.
- Onkodin – Daten und Informationen zu Onkologie und Hämatologie
- Kompetenznetz Akute und chronische Leukämien (KNL): Akute myeloische Leukämie (AML)