
| Klassifikation nach ICD-10 | |
|---|---|
| Q77.6 | Chondroektodermale Dysplasie Inkl.: Ellis-van-Creveld-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Das Ellis-van-Creveld-Syndrom (EVC), auch als chondroektodermale Dysplasie bezeichnet, ist eine sehr seltene autosomal-rezessiv vererbte Krankheit. Es gehört zur Gruppe der Kurzripp-Polydaktylie-Syndrome (SRPS für Short Rib-Polydactyly Syndrome).
Klinisches Bild



Das Ellis-van-Creveld-Syndrom ist eine chondrale und ektodermale Dysplasie (chondroektodermale Dysplasie). Sie ist bei den betroffenen Patienten durch kurze Rippen, Polydaktylie (Vielfingerigkeit), Kleinwuchs, ektodermale Defekte und Herzfehler gekennzeichnet.
Die äußerlichen Hauptsymptome sind der Kleinwuchs, Thoraxdeformität mit verkürzten Rippen, die Polydaktylie, sowie hypo- oder dysplastische Fingernägel und Zähne. Die Polydaktylie ist an den Händen üblicherweise beidseitig, postaxial (Außen) auf der ulnaren Seite. Bei etwa 10 % der Patienten sind auch die Füße von der Polydaktylie betroffen. Häufig ist zwischen der Großzehe (Hallux) und der zweiten Zehe ein größerer Zwischenraum. Die Extremitäten wirken meist plump. Die mesomele (mittlere Anteile, das heißt Unterarm beziehungsweise -schenkel) und akromele (distale Anteile, das heißt Finger beziehungsweise Zehen) Verkürzung der Gliedmaßen ist sehr häufig. Sehr oft können die Patienten die Hand nicht zu einer Faust ballen.
Bei etwa 60 % der Patienten liegt ein Herzfehler, meist in Form eines Atriumseptumdefektes (ASD), vor. Die kognitive und motorische Entwicklung verläuft dagegen normal.
Häufigkeit
Die weltweite Häufigkeit des Ellis-van-Creveld-Syndroms wird bei Neugeborenen auf etwa 1 : 60.000 bis 1 : 200.000 geschätzt. In einigen Populationen tritt es signifikant häufiger auf. So beispielsweise in der Gemeinschaft der Amischen in Lancaster County (Pennsylvania) und den Aborigines in Westaustralien. Bei den Amischen in Lancaster County liegt die Prävalenz bei etwa 1 : 5000 pro Neugeburt. Dort wurden bis zum Jahr 2000 insgesamt 52 Fälle in 30 Sippschaften berichtet.
Weltweit gab es im Jahr 2007 etwa 150 dokumentierte Fälle von EVC. Andere Quellen berichten von 300 Fällen im Jahr 1998.
Genetik und Pathologie
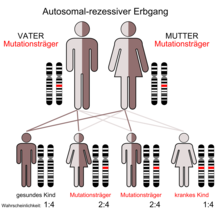
Das Ellis-van-Creveld-Syndrom wird autosomal-rezessiv vererbt. Die Ursache sind Mutationen in den Genen EVC1 (auch EVC genannt) und EVC2. Die beiden Gene liegen beim Menschen auf Chromosom 4, „Kopf an Kopf“ auf dem Genlocus p16. Andere genetische Faktoren scheinen ebenfalls einen Einfluss auf die Krankheit zu haben.
Das EVC-Gen wurde 1995 zunächst auf dem kurzen Arm von Chromosom 4 gefunden. Dies ist ein Gebiet, in dem auch andere kongenitale Chondrodystrophien ihren Ursprung haben. Mutationen in diesem Gen konnten bei EVC-Patienten in Lancaster County, sowie Mittel- und Südamerika gefunden werden. Allerdings konnte bei einem Screening von 58 EVC-Patienten nur bei 13 eine homozygote Mutation im EVC-Gen lokalisiert werden. Bei den restlichen 45 Patienten lag keine Mutation in einem oder beiden Allelen vor. Ein zweites Gen wurde bei diesen Patienten als Auslöser des Ellis-van-Creveld-Syndroms identifiziert: EVC2. Es liegt ebenfalls auf Chromosom 4, „Kopf an Kopf“ in unmittelbarer Nachbarschaft zu EVC1. Die beiden Gene sind durch lediglich 2,6 kB getrennt.EVC2 ist 166,4 kB lang und teilt mit EVC1 eine gemeinsame Promotor-Region, während die beiden Sequenzen keine signifikanten Sequenzübereinstimmungen aufweisen – weder auf Protein- noch auf Nukleinlevel. Dennoch lässt sich der Phänotyp beim Ellis-van-Creveld-Syndrom nicht anhand des mutierten Gens – EVC1 oder EVC2 – unterscheiden.
Bei der Sequenzierung von EVC1 und EVC2 bei 65 Patienten mit Ellis-van-Creveld-Syndrom wurde in 20 Fällen eine EVC1-Mutation (31 %) in beiden Allelen und in 25 Fällen eine EVC2-Mutation (38 %) gefunden. Bei letzteren waren in 22 Fällen beide Allele betroffen und in 3 nur ein Allel. In der Mehrzahl aller Fälle ist ein falsch gesetztes Stopcodon der Auslöser der Krankheit. Bei den restlichen 20 Patienten (31 %) wurde in keinem der beiden Gene eine Mutation gefunden. Es wird daher vermutet, dass das Ellis-van-Creveld-Syndrom eine heterogene genetische Erkrankung ist. Heterozygote Mutationen in EVC1 oder EVC2 lösen auch das Weyers-Syndrom, eine akrofaziale Dysostose (Knochenbildungsstörung der Hände und des Gesichts), aus, das allerdings autosomal-dominant vererbt wird. Auch dass in einigen Fällen heterozygote Merkmalsträger erkranken können – diese Patienten haben nur ein defektes EVC1- oder EVC2-Gen in jeder Körperzelle – spricht für eine komplexe Genetik, in der möglicherweise noch weitere Mutationen in anderen Genen eine Rolle spielen.

Diagnose
In der Pränataldiagnostik können mittels Feinultraschall der enge Thorax, die Verkürzung der langen Röhrenknochen, Hexadaktylie und Herzfehler ab dem späten ersten Trimester erkannt werden.
Radiologische Kriterien sind:
- Beckendysplasie mit Spornbildung medial am Pfannendach
- Vorzeitige Ossifikation der proximalen Oberarm- und Oberschenkelepiphyse
- Überproportional kurze Unterarme und Unterschenkel
- Verbreiterung der Tibia proximal, nach medial verlagertes Ossifikationszentrum mit massivem Genu valgum
- zu kurze Fibula
- Verkürzte Fingerglieder mit Zapfenepiphysen der Mittel- und Endphalangen
Häufig Hexadaktylie, Fusion von Handwurzelknochen (Os hamatum und Os capitatum) Häufig Radiusköpfchenluxation und Patellaluxation, postaxiale Polydaktylie.
Differentialdiagnose
In der Differentialdiagnose sind das Jeune-Syndrom, das McKusick-Kaufman-Syndrom, das Weyers-Syndrom, das Pallister-Hall-Syndrom und vor allem das Verma-Naumoff-Syndrom abzugrenzen, ferner die Kranioektodermale Dysplasie.
Diagnosesicherheit bietet ein Gentest auf EVC beziehungsweise EVC2.
Therapie
Das Ellis-van-Creveld-Syndrom ist derzeit nicht heilbar.
Die Patienten brauchen schon ab der Geburt eine multidisziplinäre fachärztliche Betreuung. Der enge Thorax verursacht in vielen Fällen Atemprobleme. Mögliche Herzfehler müssen überwacht und symptomatisch behandelt werden. Die Knochendeformationen erfordern eine regelmäßige orthopädische Kontrolle. Die Zähne bedürfen einer fachzahnärztlichen Betreuung. Für das perioperative / anästhesiologische Management sind aktuelle Handlungsempfehlungen erschienen (Eintrag bei OrphanAnesthesia).
Prognose
Die Prognose wird in den ersten Lebensmonaten durch die wegen des verengten Thorax eingeschränkte Atmung bestimmt. Später sind etwaige Herzfehler der wesentliche prognostische Faktor. Die adulte Körpergröße ist nur schwer vorauszusagen.
Erstbeschreibung
1940 beschrieben die beiden Pädiater Richard Ellis (1902–1966) und Simon van Creveld (1894–1971) bei drei Patienten erstmals ein Syndrom, das später nach den beiden Erstbeschreibern benannt wurde. Das Syndrom wurde teilweise bereits in einigen früheren Veröffentlichungen beschrieben. Ellis und van Creveld definierten es jedoch als erste.
Weiterführende Literatur
- S. Mehndiratta, A. Tyagi, V. Devgan: Ellis-van Creveld syndrome: report of two cases. In: World journal of pediatrics. Band 7, Nummer 4, November 2011, S. 368–370, ISSN 1867-0687. doi:10.1007/s12519-011-0256-x. PMID 21210265.
- K. Hegde, R. M. Puthran u. a.: Ellis van Creveld syndrome–a report of two siblings. In: BMJ Case Reports. 2011 doi:10.1136/bcr.09.2011.4774
- D. Alves-Pereira, L. Berini-Aytés, C. Gay-Escoda: Ellis-van Creveld syndrome. Case report and literature review. (PDF; 334 kB) In: Medicina oral, patología oral y cirugía bucal. Band 14, Nummer 7, Juli 2009, S. E340–E343, ISSN 1698-6946. PMID 19300361. (Review).
- M. Weber, A. Kaufmann u. a.: Orthopädische Probleme beim Ellis-van-Creveld-Syndrom. In: Monatsschrift Kinderheilkunde. Band 149, Nummer 1, 2001, S. 51–57. doi:10.1007/s001120050724
- M. G. Blackburn, R. E. Belliveau: Ellis-van Creveld syndrome. A report of previously undescribed anomalies in two siblings. In: American Journal of Diseases of Children. Band 122, Nummer 3, September 1971, S. 267–270, ISSN 0002-922X. PMID 5568596.
Weblinks
- Ellis-van Creveld Syndrome; EVC. In: Online Mendelian Inheritance in Man. (englisch)
- EVC Gene. In: Online Mendelian Inheritance in Man. (englisch)
- EVC2 Gene. In: Online Mendelian Inheritance in Man. (englisch)
- Ellis-van-Creveld-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Ellis-van Creveld syndrome (englisch)