
| Klassifikation nach ICD-10 | |
|---|---|
| D12 | Gutartige Neubildung des Kolons, des Rektums, des Analkanals und des Anus |
| D12.6 | Kolon, nicht näher bezeichnet Polyposis coli (hereditär) |
| ICD-10 online (WHO-Version 2019) | |
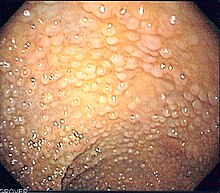
Die familiäre adenomatöse Polyposis (FAP) ist eine autosomal-dominant vererbte Erkrankung, bei der es zu einem massenhaften Befall des Dickdarms mit Polypen kommt. Unbehandelt liegt die Wahrscheinlichkeit der Entartung eines oder mehrerer dieser Polypen zu Darmkrebs bei nahezu 100 %.
Häufigkeit
Die Erkrankung ist selten. Es wird geschätzt, dass 5–10 von 100.000 Menschen von der Genmutation betroffen sind.
Ursache
Ursache der Erkrankung ist eine Keimbahnmutation des APC-Gens (Adenomatous Polyposis of the Colon). Eines der beiden Allele dieses auf Chromosom 5 5q21-q22 liegenden Tumorsuppressorgens ist dabei inaktiviert. Kommt nun noch eine weitere (somatische) Mutation hinzu, kann auch das andere Allel inaktiviert werden. Beim Gesunden bindet APC-Protein an β-Catenin und vermittelt über Ubiquitinylierung dessen Abbau im Proteasom. Kann β-Catenin nicht mehr ubiquitinyliert werden, enthalten die Zellen dadurch große Mengen an β-Catenin. Dieses transloziert in den Zellkern und wirkt dort gemeinsam mit anderen Faktoren als Transkriptionsfaktor. Dadurch wird das Zellwachstum gefördert und Telomerase gebildet, die Zellen immortalisiert (den Zelltod verhindert). Es kommt zu einem beschleunigten Ablauf der Adenom-Karzinom-Sequenz.
Klinische Erscheinungen
Die ersten Dickdarm-Polypen treten bei FAP-Patienten in der Regel zwischen dem 10. und 25. Lebensjahr auf. Die Krankheit kann zunächst über mehrere Jahre unbemerkt verlaufen. Zu einem späteren Zeitpunkt können folgende Beschwerden (Symptome) auftreten:
- Blut- und/oder Schleimabgang aus dem Darm
- Durchfälle oder Verstopfung, oder häufiger Wechsel zwischen beidem
- Blähungen
- Schmerzen im Bauch oder im Enddarmbereich
- Gewichtsverlust
Neben der klassischen FAP gibt es auch eine mildere Variante, die so genannte attenuierte FAP oder AFAP. Diese ist durch ein späteres Erkrankungsalter und in der Regel weit weniger Polypen (< 100) im Dickdarm gekennzeichnet. Trotz des eher milderen Verlaufs ist das Lebenszeitrisiko für Dickdarmkrebs aber ähnlich hoch wie bei der klassischen FAP. Bei einigen Patienten werden auch – meist gutartige – Veränderungen außerhalb des Dickdarms beobachtet. Manchmal treten diese Erscheinungen bereits vor den Dickdarmpolypen auf. Die Beobachtung der im Folgenden genannten Veränderungen kann auf das Vorliegen einer FAP hinweisen und sollte immer Anlass zu weitergehenden Untersuchungen sein.
Bei einer autosomal-dominant vererbten Erkrankung entsteht das entsprechende Erkrankungsbild bereits dann, wenn nur eines der beiden paarig angelegten Gene verändert (mutiert) ist. Da man an seine Kinder nur jeweils ein Gen eines Genpaares weitergibt, beläuft sich das Risiko für Kinder eines Erkrankten, das veränderte Gen zu erben, auf jeweils 50 %. Beim autosomal-dominanten Erbgang spielt das Geschlecht bei der Vererbung keine Rolle, das heißt, sowohl Männer als auch Frauen können die Veränderung geerbt haben bzw. weitervererben.
Wenn bei einem Betroffenen die verantwortliche Mutation identifiziert wurde, ist es möglich, alle Angehörigen einer Familie im Rahmen einer humangenetischen Beratung auf das Vorliegen dieser Mutation zu testen, bevor erste klinische Symptome beobachtet werden (vorhersagende bzw. prädiktive Diagnostik). Für nachgewiesene Mutationsträger (Anlageträger) besteht ein praktisch 100-prozentiges Erkrankungsrisiko, sie sollten deshalb intensive Vorsorge- bzw. Früherkennungs-Untersuchungen und gegebenenfalls therapeutische Maßnahmen hinsichtlich einer FAP durchführen lassen.
Untersuchungsmethoden
Als frühestes Merkmal der Erkrankung ist bei der Geburt vorhandene Vermehrung des Pigmentepithels der Regenbogenhaut erkennbar. Zur sicheren Diagnosestellung sollten die Familienmitglieder per Darmspiegelung untersucht werden, da sie ebenso das typische Bild einer Vielzahl von Polypen zeigen.
Therapie und Verlauf
Die bisher einzige Möglichkeit, das Auftreten von Dickdarmkrebs bei Menschen mit einer klassischen FAP zu verhindern, besteht in der operativen Entfernung des Dickdarms (Kolektomie). Diese Operation kann heute fast immer kontinenzerhaltend durchgeführt werden. Der Zeitpunkt für die Operation sollte individuell entschieden werden. In den allermeisten Fällen führen die betroffenen Patienten nach der Operation wieder ein unbeeinträchtigtes soziales, berufliches und sexuelles Leben. Es ist jedoch wichtig, dass FAP-Patienten an Zentren betreut und operiert werden, in denen große Erfahrung mit der Behandlung dieser Erkrankung besteht. Die Behandlung eines betroffenen FAP-Patienten orientiert sich in der Regel nicht am Ergebnis der molekulargenetischen Diagnostik, sondern am klinischen Verlauf, das heißt an Zahl, Wachstumsgeschwindigkeit und feingeweblichem Befund der Polypen.
Neben der chirurgischen Behandlung kann bei einigen Patienten mit milden Verlaufsformen der FAP oder bei erhaltenem Enddarm nach Kolektomie auch ein medikamentöser Therapieversuch besprochen werden: Mit dem Medikament Sulindac kann das Polypenwachstum bei manchen FAP-Patienten reduziert werden. Eine chirurgische Behandlung kann hierdurch allerdings in vielen Fällen – insbesondere bei einem stark zunehmenden Polypenwachstum – nicht vermieden werden.
Polypen im Zwölffingerdarm (Duodenum): das Lebenszeitrisiko für das Auftreten von Adenomen liegt bei 80–90 %. Polypen im Zwölffingerdarm verursachen meist keine Beschwerden. Dennoch können solche Adenome – wenngleich nicht so häufig wie im Dickdarm – durch Größenwachstum bei etwa 4–12 % der FAP-Patienten bösartig werden (Duodenal-Karzinom). Deshalb sind regelmäßige Untersuchungen (Magen-Zwölffingerdarm-Spiegelung) unbedingt zu empfehlen.
Polypen im Magen: hierbei handelt es sich meist um gutartige Drüsenkörperzysten, die keiner weiteren Behandlung bedürfen (bei etwa 30 % der Patienten), seltener (circa 10 %) werden Magen-Adenome festgestellt. Das Risiko für Magenkrebs bei FAP-Patienten ist allenfalls geringfügig erhöht (vermutlich circa 0,5 % der Betroffenen).
Desmoide: bindegewebige Tumoren, die vor allem postoperativ in der Bauchwand oder im Bauchraum entstehen. Sie bilden zwar keine Metastasen, können unter Umständen aber sehr groß werden und dann auch auf andere Organe verdrängend wirken (bei 10–30 % der Patienten).
Pigmentflecken der Netzhaut (congenitale Hypertrophie des retinalen Pigment-Epithels = CHRPE): nur durch eine augenärztliche Untersuchung nachweisbare harmlose dunkle Flecken der Netzhaut, die das Sehvermögen nicht beeinträchtigen (bei etwa 80 % der Patienten mit klassischer FAP).
Epidermoidzysten: gutartige Geschwulste unter der Haut, die häufig bereits im Kindesalter auftreten (bei etwa 50 % der Patienten).
Zahnanomalien: Unregelmäßigkeiten der Zahnform oder Zahnzahl (bei etwa 15–20 % der Patienten).
Osteome: gutartige Knochentumoren, vor allem am Kiefer, im Gesicht und am Schädel (bei 75–90 % der Patienten).
Schilddrüsenkrebs: papilläre Schilddrüsenkarzinome treten vermutlich bei circa 2 % der Betroffenen auf, meist zwischen dem 15. und 30. Lebensjahr.
Medulloblastome: hierbei handelt es sich um einen Hirntumor, der typischerweise im Kindes- oder Jugendalter im Bereich der hinteren Schädelgrube entsteht (vermutlich bei weniger als 1 % der Patienten).
Hepatoblastome: hierbei handelt es sich um einen embryonalen Tumor der Leber, der bevorzugt in den ersten fünf Lebensjahren auftritt (bei circa 1 % der Betroffenen).
Attenuierte familiäre adenomatöse Polyposis (aFAP)
Die aFAP ist eine besondere Form der FAP. Sie ist eine mildere Form, bei der sich die klinische Symptomatik nicht so schnell entwickelt. Als Ursache werden Mutationen des APC-Gens, aber auch des MUTYH-Gens gefunden. Bei Patienten mit aFAP kommt es durchschnittlich erst ab dem 31. Lebensjahr zu Symptomen. Polypen sind ebenfalls weniger vorhanden und beschränken sich auf eine Anzahl von unter 100. Meist sind diese Polypen im aufsteigenden Dickdarm (Colon ascendens) zu finden.
Siehe auch
Literatur
- Horst-Dieter Becker, Werner Hohenberger, Theodor Junginger, Peter Michael Schlag (Hrsg.): Chirurgische Onkologie. Thieme, Stuttgart 2002, ISBN 3-13-126111-0.
- E. J. Gardner: A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum. In: Am J Hum Genet. 1951;3, S. 167–176. PMID 14902760
- Polymnia Galiatsatos, William D. Foulkes: Familial Adenomatous Polyposis. In: American Journal of Gastroenterology. 2006, Am. Coll. of Gastroenterology, ISSN 0002-9270, S. 385–398.
- V. Fendrich, D. K. Bartsch: Hereditäre gastrointestinale Neoplasien. In: Z Gastroenterol. 2005; 43, S. 219–225, Georg Thieme Verlag, Stuttgart/ New York, ISSN 0044-2771