
| Strukturformel | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
| Allgemeines | ||||||||||||||||||||||
| Freiname | Fludeoxyglucose (18F) | |||||||||||||||||||||
| Andere Namen |
|
|||||||||||||||||||||
| Summenformel | C6H11FO5 | |||||||||||||||||||||
| Externe Identifikatoren/Datenbanken | ||||||||||||||||||||||
| ||||||||||||||||||||||
| Arzneistoffangaben | ||||||||||||||||||||||
| ATC-Code | ||||||||||||||||||||||
| Eigenschaften | ||||||||||||||||||||||
| Molare Masse | ||||||||||||||||||||||
| Aggregatzustand |
fest |
|||||||||||||||||||||
| Schmelzpunkt |
170–176 °C |
|||||||||||||||||||||
| Sicherheitshinweise | ||||||||||||||||||||||
| ||||||||||||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | ||||||||||||||||||||||

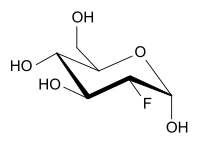
2-Fluor-2-desoxy-D-glucose (vereinfacht Fluordesoxyglucose, kurz FDG) ist ein Strukturanalogon des Einfachzuckers D-Glucose. An der Position 2 befindet sich anstelle einer OH-Gruppe ein Fluoratom.
Es wird als In-vivo-Diagnostikum in der Medizin verwendet. Der natürliche Zucker und sein Mimetikum FDG werden von den Zellen des menschlichen Körpers zunächst in gleicher Weise aufgenommen, aber anschließend unterschiedlich verstoffwechselt. Dies hat eine bezweckte Anreicherung des Diagnostikums in bestimmten Körperzellen zur Folge. In den zu untersuchenden Geweben wird die Konzentration des FDG-Tracers tomographisch erfasst. Auf diese Weise werden Normabweichungen lokalisiert und damit Hinweise auf organische Funktionsstörungen erhalten. Mit dem Radionuklid Fluor-18 (18F) markierte FDG ist das am häufigsten verwendete Radiopharmakon in der Positronen-Emissions-Tomographie. Strahlungsfreie FDG wird experimentell in der Magnetresonanztomographie erprobt. Als Therapeutikum ist FDG nicht zugelassen.
Geschichte
Die Synthese von Fluordesoxyglucose wurde erstmals im Jahr 1968 von Josef Pacák, Zdeněk Točík und Miloslav Černý an der Karls-Universität in Prag entwickelt. Im Jahr 1970 wurde ein alternativer Syntheseweg veröffentlicht. Die Eigenschaft von FDG, das für den Zuckerstoffwechsel bedeutsame Enzym Hexokinase zu hemmen, wurde 1972 beschrieben. Tatsuo Ido, Alfred P. Wolf und Joanna Fowler vom Brookhaven National Laboratory beschrieben 1977 als Erster die Synthese von radioaktivem 18F-FDG. Dabei gelang die erste Synthese Tatsuo Ido in der Gruppe von Wolf. Diese Verbindung wurde von Abass Alavi im August jenes Jahres an der University of Pennsylvania zwei Freiwilligen injiziert. Erste Aufnahmen des Gehirns, bei denen die Verteilung von FDG in diesem Organ erstmals dargestellt wurde, wurden mit einer gewöhnlichen Gammakamera durchgeführt, nicht mit einem Positronen-Emissions-Tomographen. 1984 wurde ein Verfahren beschrieben, das, nach einer Weiterentwicklung im Jahr 1986, seither als Vorlage für die Herstellung des Radiopharmakons dient. Heute wird es in der Onkologie und Neurologie verwendet, wo es das mit Abstand am häufigsten verwendete Diagnostikum ist.
Synthese
Die kurze Halbwertszeit des radioaktiven Fluorisotops stellt hohe Anforderungen an die Produktion des Radiopharmakons. Rasche Abläufe von Synthese, Reinigung und Sterilisierung müssen sichergestellt sein. Heute läuft die Herstellung von 18F-2-Fluor-2-desoxy-D-Glucose automatisiert ab. Dem Umfang der Qualitätskontrolle sind durch die Umstände Grenzen gesetzt.
Für die Herstellung sind eine Reihe von möglichen Reaktionen beschrieben. Hauptsächlich gibt es zwei Synthesewege: die elektrophile Addition, das heißt die Addition von 18F-F2 an Doppelbindungen, und die nukleophile Substitution mit 18F−.
Nukleophile Substitution
Der Weg über die nukleophile Substitution ist der Routineweg und sei hier exemplarisch beschrieben, wobei in den verschiedenen Verfahren der nukleophilen Substitution die Schutzgruppen entweder mit Säuren oder Basen entfernt werden.
Herstellung von 18F−
Das natürliche Fluoratom mit der Nukleonenzahl 19 ist im 2-FDG-Molekül in diesem Fall durch das radioaktive Fluor-18-Isotop ersetzt worden. Dieses Isotop ist ein Positronen-Emitter mit einer Halbwertszeit von lediglich 109,8 Minuten. Es ist aufgrund des schnellen Zerfalls in der Natur nicht vorhanden. Zur Herstellung von 18F-2-FDG wird dieses Isotop meist mit Hilfe eines Zyklotrons gewonnen, beispielsweise durch Beschuss des schweren Sauerstoff-Isotops 18O mit Protonen. Die Herstellung aus 20Ne mittels Deuteronen-Beschuss wurde 1986 zur Herstellung des ersten zu Forschungszwecken hergestellten FDG angewendet, liefert jedoch geringere Ausbeuten.
In einem Zyklotron wird üblicherweise ein Target aus mit dem Sauerstoffisotop 18O angereichertem Wasser H218O mittels eines Protonenzyklotrons mit Protonen (10 bis 15 MeV) beschossen. Dabei wird in einer Kernreaktion ein kleiner Teil des 18O-Sauerstoffs unter Aufnahme je eines Protons und der Abgabe eines Neutrons in das radioaktive Fluorisotop 18F umgewandelt.

Anschließend muss das radioaktive Isotop getrennt und chemisch weiterverarbeitet werden.
Umsetzung mit einem Präkursor

Über einen Ionenaustauscher wird das gebildete Fluorid vom Wasser abgetrennt und anschließend mit einer Lösung aus Acetonitril, Kryptofix 222 und Kaliumcarbonat vom Ionenaustauscher wieder abgetrennt (eluiert) (2).
In der eigentlichen nukleophilen Substitution ersetzt das radioaktive Fluorid-Ion eine leicht zu entfernende Abgangsgruppe, wie beispielsweise ein Triflat. Ein geeigneter Präkursor für die Herstellung von 2-FDG mittels nukleophiler Substitution ist 1,3,4,6-O-Acetyl-2-O-trifluormethansulfonyl-β-D-mannopyranose (1), kurz als Mannose-Triflat bezeichnet. Nach der Substitution des Triflats werden die vier Acetyl-Schutzgruppen mittels basischer oder saurer Hydrolyse mit verdünnter Natronlauge bzw. verdünnter Salzsäure entfernt (4).
Elektrophile Addition
Daneben existiert der Syntheseweg der Addition mit 18F-F2. Durch eine Reaktion von 3,4,5-tri-O-Acetyl-D-glucal entstehen zwei Produkte, 2-FDG und 2-Fluordesoxymannose:

Aufreinigung
Durch eine Festphasenextraktion oder eine Umkehrphasen-Hochleistungsflüssigkeitschromatographie (RP-HPLC) kann das gebildete 2-FDG sauber von den Ausgangsstoffen abgetrennt werden. Die Aufreinigung ist ein wichtiger Schritt bei der Herstellung von 2-FDG, um den Vorgaben der einzelnen Pharmakopöen und des Good Manufacturing Practice (GMP) zu entsprechen. Nach dem europäischen Arzneibuch muss mehr als 95 % der Radioaktivität der Fluordesoxyglucose und der Fluordesoxymannose entstammen, wobei der Anteil der Fluordesoxymannose maximal 10 % der Radioaktivität betragen darf. Die Aufreinigungsschritte finden in geschlossenen und mit Bleizellen abgeschirmten Geräten statt.
Metabolismus
18F-Fluordesoxyglucose wird von den Zellen des menschlichen Körpers wie Glucose aufgenommen, obwohl an einer Stelle des Moleküls eine Hydroxygruppe durch das Radionuklid 18F ersetzt ist. Dabei wird 2-FDG von den Zellen mittels Glucosetransporter aus dem Blut aufgenommen. Im Rahmen des Humangenomprojekt wurden 14 Glucosetransporter (GLUT) bei Menschen identifiziert.
GLUT-1 ist das wichtigste Transportprotein für die Aufnahme von 2-FDG in Tumoren und normalem Hirngewebe. Die Aufnahme in der Skelettmuskulatur und im Herzmuskel ist durch Insulin stimulierbar und erfolgt über den GLUT-4-Transporter. Das Enzym Hexokinase phosphoryliert 2-FDG anschließend innerhalb der Zelle. 2-FDG kann allerdings von den Zellen nach der Phosphorylierung nicht weiter verstoffwechselt werden. Die Rückreaktion, die Dephosphorylierung von FDG-6-Phosphat zu FDG, erfolgt in allen Organen – mit Ausnahme der Leber – und im Tumorgewebe sehr langsam. Deshalb findet eine Anreicherung von FDG-6-Phosphat in den Zellen statt (metabolic trapping). Anhand des Zerfalls von 18F kann 2-FDG detektiert werden. Die Verteilung von 2-FDG im Körper erlaubt Rückschlüsse auf den Glucosestoffwechsel verschiedener Gewebe. Dies ist besonders für die frühe Diagnose von Krebserkrankungen von Vorteil, da eine Tumorzelle typischerweise aufgrund eines erhöhten Stoffwechsels viel Glucose verbraucht und dementsprechend 2-FDG anreichert. Die Stoffwechselaktivität eines Tumors wird mit Hilfe des SUV-Wertes quantitativ beschrieben.
Als ein der Glucose sehr ähnliches Molekül überwindet 2-FDG problemlos die Blut-Hirn-Schranke. Da das menschliche Gehirn einen hohen Bedarf an Glucose hat, wird ein entsprechend großer Anteil von 2-FDG im Gehirn angereichert. Des Weiteren reichert sich beim gesunden Menschen 2-FDG in den Nieren und in den ableitenden Harnwegen an.
Das 18F zerfällt zu 18O, einem natürlichen Sauerstoffisotop. Unmittelbar nach dem Zerfall bildet sich nach Aufnahme eines freien Wasserstoffatoms aus der Umgebung eine Hydroxygruppe.
Die gebildete Glucose wird anschließend auf normalem Wege in der Glykolyse metabolisiert, während 18F-2-FDG-Phosphat das Enzym des folgenden Reaktionsschrittes der Glykolyse hemmt (die Glucose-6-phosphat-Isomerase). Tumorzellen besitzen auch nicht ausreichende Mengen des Enzyms Glucose-6-Phosphatase, um die Rückreaktion von 18F-2-FDG-Phosphat zu 18F-2-FDG zu katalysieren.
Die durch den Zerfall von 18F-2-FDG mögliche Bildgebung ist ein Abbild der Verteilung der Glucose-Aufnahme und -Phosphorylierung der Zellen im menschlichen Körper. FDG zeigt auch eine erhöhte Aufnahme in Tumoren von Mäusen, Ratten, Hamstern und Kaninchen. In Ratten zeigt 2-FDG in Tumoren aufgrund der erhöhten Stoffwechselaktivität eine Stunde nach Injektion eine Anreicherung auf das 22-Fache im Vergleich zum Blutkreislauf, die für eine weitere Stunde konstant bleibt.
Anwendungen


2-FDG wird in der PET für die Diagnose,Staging (Stadienbestimmung), Therapieeinstellung und Therapiekontrolle verwendet. Man spricht in diesem Zusammenhang auch oft von der „FDG-PET“. 2-FDG ist als Diagnostikum eine außerordentlich nützliche und vielfach bewährte Verbindung. Die Anwendung hat einen rein diagnostischen Hintergrund. Genutzt wird dabei die bei der Paarvernichtung (Annihilation) von Positron und Elektron entstehende Vernichtungsstrahlung. Bei der Annihilation entstehen zwei hochenergetische Photonen, die eine Energie von 511 keV haben und in einem Winkel von 180 Grad zueinander, ausgesandt werden. Für die Therapie (Strahlentherapie, in diesem besonderen Fall würde man von einer Endoradiotherapie sprechen) sind die für die Diagnostik genutzten Gammaquanten nicht geeignet.
Neben der Hauptanwendung in der Onkologie wird 2-FDG auch für die Diagnose der Alzheimerschen Krankheit, der Parkinsonschen Krankheit, von Epilepsien, in der Kardiologie und in der Entzündungsdiagnostik verwendet, allerdings in weit geringerem Maße als in der Onkologie.
Für die FDG-PET gibt es drei Hauptindikationen für die Untersuchung von Patienten mit onkologischen Erkrankungen: die Differenzierung zwischen benignen oder malignen (gutartig oder bösartig) Tumoren, das Tumorstaging bezüglich der Lymphknoten und Fernmetastasen sowie die Differenzierung Narbengewebe/vitales Tumorgewebe (Rezidiv, residueller Tumor). Die FDG-PET wird in der Onkologie zur Untersuchung von Lungenkrebs, dem kolorektalem Karzinom, Speiseröhrenkrebs, Magenkrebs, Kopf-Hals-Karzinom, Gebärmutterhalskrebs, Eierstockkrebs, Brustkrebs, dem malignen Melanom und den meisten Arten von Lymphomen eingesetzt. Insbesondere sehr langsam wachsende Tumoren weisen in der Regel keine wesentlich erhöhte FDG-Aufnahme aus. Eine FDG-PET-Untersuchung ist dann meist nur in Ausnahmefällen sinnvoll. Dazu gehören Prostatakarzinome, differenzierte neuroendokrine Tumoren (z. B. Karzinoid), bronchoalveoläre Karzinome, niedrig maligne Non-Hodgkin-Lymphome, niedrig maligne Hirntumore (Astrozytom II, Oligodendrogliom II) und das Leberzellkarzinom (vor allem höher differenzierte Formen). Entzündungen bzw. Heilungen zeigen neben dem Tumorgewebe ebenfalls eine erhöhte Stoffwechselaktivität und somit eine erhöhte FDG-Aufnahme. Eine Untersuchung zur Differenzierung beispielsweise von Abszessen und Tumorgewebe, Sarkoidose, Bronchialkarzinomen usw., kann deshalb mit 2-FDG kaum sinnvoll durchgeführt werden.
Hohe Blutzuckerspiegel führen zu einer reduzierten Aufnahme von 2-FDG im Tumorgewebe, was das Signal-Rausch-Verhältnis verschlechtert. Bei Nüchternblutzuckerwerten über 150 mg/dl wird deshalb die Indikation zur FDG-PET meist kritisch überprüft. Nach Abschluss einer Chemo- oder Strahlentherapie kommt es auch bei vitalen Tumorzellen häufig zu einer Reduktion der FDG-Aufnahme. Deshalb sollte zwischen PET-Untersuchung und Abschluss der Therapie ein Zeitraum von mindestens vier Wochen liegen. Eine Ausnahme sind Verlaufsuntersuchungen bzw. bestimmte klinische Studien. Alternativen zu 2-FDG sind z. B. radiomarkierte Aminosäuren wie 11C-Methionin zur Bestimmung der Proteinbiosynthese, radiomarkiertes 11C-Cholin zur Bestimmung der Synthese der Membranlipide und radiomarkiertes 11C-Acetat zur Bestimmung der Fettsäuresynthese. Jedoch ist in den USA nur 2-FDG als Tracer von der Food and Drug Administration zugelassen. In Deutschland wurde FDG 2000 und 2005 als Tracer zugelassen.
Applikation
Im Fall des Ganzkörper-Scans auf der Suche nach Tumoren oder deren Metastasen wird eine Dosis von etwa 200 bis 400 MBq über eine isotonische Kochsalzlösung in eine Vene des Patienten injiziert. Über die Körperoberfläche des Patienten wird die zu applizierende Aktivitätsmenge errechnet. Die Zielgröße ist es dabei, etwa 210.000 Ereignisse pro Schicht im PET registrieren zu können. Die empfohlene Maximaldosis einer FDG-Injektionslösung liegt bei 10 mg, entsprechend 55 Mikromol.
Der Patient muss vor der Applikation von 2-FDG mindestens für sechs Stunden nüchtern bleiben, um einen möglichst niedrigen Blutzuckerspiegel zu haben. Diese Anforderung ist für einige Diabetiker problematisch, da die entsprechenden Kliniken meist keine PET-Untersuchung durchführen, wenn der Blutzuckerspiegel über 10 mmol/l liegt. Der venöse Blutzuckerspiegel wird vor jeder FDG-PET-Untersuchung bestimmt.
Nach erfolgter Injektion muss der Patient meist für eine Stunde in möglichst völliger Ruhe ohne körperliche Betätigung liegen, um die gleichmäßige Verteilung von 2-FDG im Körper zu gewährleisten. Muskuläre Anstrengungen würden 2-FDG zu den entsprechenden Muskeln leiten und das Ergebnis verfälschen bzw. zu Artefakten bei der Bildgebung führen. Oft beobachtet man im Bereich der Zunge eine starke Anreicherung von 2-FDG, was durch häufige und starke Schluckbewegungen der zum Teil unter extremem psychischen Stress stehenden Patienten bedingt ist.
Vor der Applikation können Wasser oder andere kalorienfreie Getränke vom Patienten zu sich genommen werden. Unmittelbar vor Beginn der Aufnahme soll der Patient die Blase entleeren.
Strahlenexposition
Die Strahlendosis bei einer PET mit 2-FDG beträgt ca. 7–10 mSv. Im Vergleich dazu beträgt die Strahlendosis bei einer kontrastverstärkten Computertomographie ca. 20–40 mSv. Die Höhe jener Dosis entspricht etwa der doppelten bis dreifachen Dosis der natürlichen Strahlenexposition, der die europäische Bevölkerung jährlich im Durchschnitt ausgesetzt ist (ca. 3 mSv pro Jahr). Das Risiko des Auftretens von Nebenwirkungen durch die Strahlung ist aus diesem Grunde vernachlässigbar klein. Die effektive Dosis nach der intravenösen Injektion von 2-FDG beträgt 2,0 × 10−2 mSv/MBq. Die höchste Strahlenexposition liegt dabei für die Harnblase bei 1,7 × 10−1 mSv/MBq. Nach einer Applikation soll für mindestens 10 Halbwertszeiten (1098 Minuten, entsprechend etwa 18 Stunden) nicht gestillt werden.
Nebenwirkungen
Es sind keine allergischen oder toxischen Nebenwirkungen bekannt. Die extrem geringe Dosis an injiziertem 2-FDG, die im Bereich Pikomol bis Nanomol liegt, und die vergleichsweise milde Strahlungsart schließen dies aus. Im Vergleich dazu bewegen sich die in der CT oder bei der Magnetresonanztomographie verwendeten Mengen an Kontrastmitteln im Bereich von einigen Millimol, das heißt, sie liegen um etwa 6 bis 9 Größenordnungen höher.
Therapeutische Anwendungen
Während die Gammastrahlung von 2-FDG therapeutisch weitgehend nutzlos ist, hat die β+-Strahlung des Positrons, ähnlich der β−-Strahlung eines Elektrons, durchaus therapeutisches Potenzial. Dies ergibt sich vor allem durch die geringe Reichweite der Positronen im Tumorgewebe – sie liegt bei lediglich 1 bis 2 mm – und die recht hohe selektive Anreicherung von 2-FDG im Tumorgewebe. Es wurden erste Studien bei Melanom, Brustkrebs und kolorektalem Karzinom durchgeführt. Eine Zulassung zur Behandlung von Krebserkrankungen hat 2-FDG bisher nicht.
Literatur
- Klaus Wienhard, Rainer Wagner, Wolf-D. Heiss: PET. Grundlagen und Anwendungen der Positronen-Emissions-Tomographie. Springer, Berlin u. a. 1989, ISBN 3-540-19451-7.
- Klaus Kopka, Otmar Schober, Stefan Wagner: 18F-labelled cardiac PET tracers: selected probes for the molecular imaging of transporters, receptors and proteases. In: Basic Research in Cardiology, 2008, 103. Jahrgang, Heft 2, S. 131–143; doi:10.1007/s00395-008-0703-6; PMID 18324369.
- A. Zhu, D. M. Marcus, H. K. Shu, H. Shim: Application of Metabolic PET Imaging in Radiation Oncology. In: Radiation Research, 2012, 177. Jahrgang, Heft 4, S. 436–448; PMID 22339451; PMC 3922713 (freier Volltext).