
Die Genexpressionsanalyse ist eine Untersuchung der Umsetzung der genetischen Information (Genexpression) mit molekularbiologischen und biochemischen Methoden. Sie kann sowohl für einzelne Transkripte als auch für das ganze Transkriptom angewendet werden und ermöglicht qualitative und quantitative Aussagen über die Aktivität der Gene. Während im ersten Fall nur die Sequenzinformation des zu untersuchenden Transkripts bekannt sein muss, wird für die Transkriptom-Analyse die gesamte Sequenzinformation des Transkriptoms gebraucht.
Eigenschaften
In der Molekularbiologie kann mit Hilfe der Genexpressionsanalyse die Aktivität und Expression tausender Gene gleichzeitig gemessen werden, was einen Überblick über zelluläre Funktionen ermöglicht. Genexpressionsprofile können beispielsweise verwendet werden, um Zellen zu identifizieren, die in der aktiven Teilungsphase sind, oder aber um die Reaktion von Zellen auf eine spezielle Behandlung aufzuzeigen. Viele derartige Experimente untersuchen das gesamte Genom, d. h. jedes Gen einer spezifischen Zelle.
Die DNA-Microarray-Technik misst die relative Aktivität zuvor identifizierter Zielgene. Sequenz-basierte Methoden wie die serielle Analyse der Genexpression (SAGE, SuperSAGE) werden ebenfalls zur Genexpressionsanalyse verwendet. SuperSAGE ist besonders genau, da diese Methode nicht auf zuvor definierte Gene beschränkt ist, sondern jedes aktive Gen messen kann. Seit der Einführung von Sequenzierungsmethoden der nächsten Generation erfreut sich die Sequenz-basierte „Gesamt-Transkriptom-Shotgun-Sequenzierung“, auch RNA-Seq, zunehmender Beliebtheit, da sie eine digitale Alternative zu Microarrays darstellt. Ein Vergleich von PubMed-Artikeln des Jahres 2015 mit den Begriffen „Microarray“ bzw. „RNA-Seq“ zeigt, dass Microarrays noch etwa dreimal häufiger in den Publikationen Anwendung fanden.
Qualitative Analyse:
- Wird ein Gen unter den untersuchten Umständen überhaupt exprimiert?
- In welchen Zellen findet eine Expression statt (beispielsweise in situ Hybridisierung)?
Quantitative Analyse:
- Wie stark ist der Expressionsunterschied gegenüber einer definierten Referenz?
- gesunde vs. kranke Zellen
- Wildtyp vs. Mutantenzellen
- unstimulierte vs. stimulierte Zellen
Je nach Methode werden die Produkte der verschiedenen Ebenen der Genexpression analysiert:
Die Expressionsanalyse ist der nächste logische Schritt nach der Sequenzierung eines Genoms: die Sequenz gibt Auskunft darüber, was eine Zelle möglicherweise tun könnte, während ein Expressionsprofil zeigt, was die Zelle tatsächlich gerade tut. Gene enthalten die Information zur Erzeugung von Messenger-RNA (mRNA), aber zu jedem beliebigen Zeitpunkt schreibt eine Zelle nur einen Bruchteil ihrer Gene in mRNA um. Wenn ein Gen gerade abgelesen (exprimiert) wird, um mRNA zu produzieren, wird es als „eingeschaltet“ betrachtet, andernfalls als ausgeschaltet. Viele verschiedene Faktoren bestimmen, ob ein Gen ein- oder ausgeschaltet ist. So haben beispielsweise die Tageszeit, die lokale Umgebung, chemische Signale von anderen Zellen sowie die Frage, ob sich die Zelle gerade in aktiver Teilung befindet oder nicht, einen Einfluss auf die Aktivität der Gene. Hautzellen, Leberzellen und Nervenzellen exprimieren zu einem gewissen Grad unterschiedliche Gene, und das erklärt einen Großteil der Unterschiede dieser Zellen. Daher erlaubt eine Expressionsanalyse u. a. Schlussfolgerungen über den Zelltyp sowie den Zustand und die Umgebung einer Zelle. Expressionsanalysen untersuchen häufig die relativen Mengen an mRNA, welche unter zwei oder mehr experimentellen Bedingungen exprimiert werden. Veränderte Pegel einer spezifischen mRNA-Sequenz deuten auf eine Änderung im Bedarf des Proteins an, für welches diese mRNA codiert. Eine derartige Änderung könnte sowohl eine homöostatische Antwort als auch einen pathologischen Zustand anzeigen.
Beispiel
Ein erhöhter Pegel der für Alkoholdehydrogenase codierenden mRNA darauf hin, dass die untersuchten Zellen oder Gewebe auf eine erhöhte Alkoholkonzentration reagieren. Wenn Brustkrebszellen eine größere Menge von mRNA eines speziellen Transmembran-Rezeptors exprimieren als normale Zellen, so könnte dies ein Hinweis darauf sein, dass dieser Rezeptor eine Rolle bei Brustkrebs spielt. Ein Medikament, welches diesen Rezeptor hemmt, könnte zur Vorbeugung oder Behandlung von Brustkrebs eingesetzt werden. Bei der Entwicklung eines Medikaments können Genexpressionsanalysen dabei helfen, dessen Toxizität zu bestimmen, indem man nach Veränderungen im Expressionsniveau eines Biomarkers für die Verstoffwechselung von Medikamenten wie etwa Cytochrom P450 schaut. Genexpressionsanalysen könnten somit ein wichtiges diagnostisches Hilfsmittel werden.
Methoden
- Mit der in-situ-Hybridisierung wird sequenzspezifisch RNA eines definierten Gens/Gen-Sets im Gewebe detektiert und das lokale Genexpressionsmuster bestimmt.
RNA basiert
Nach einer RNA-Reinigung können verschiedene Methoden eingesetzt werden:
- Bei der Northern-Blot-Methode wird RNA zunächst isoliert und elektrophoretisch nach ihrer Größe in einem Gel aufgetrennt. Nach Übertragung auf eine Membran (Blotting) wird die gesuchte RNA-Sequenz durch markierte Sonden (Radioisotope, Fluoreszenz-Farbstoffe) aus komplementärer RNA oder DNA über komplementäre Bindung nachgewiesen. In der Regel werden nur geringe Anzahlen von Sequenzen simultan untersucht.
- Beim RNase Protection Assay werden RNA mit spezifisch hybridisierenden radioaktiv markierten antisense-RNA-Hybridisierungssonden vor einem Abbau durch Einzelstrang-abbauende RNasen geschützt. Die geschützten RNA-Moleküle werden mittels Gelelektrophorese aufgetrennt, autoradiographisch nachgewiesen und quantifiziert.
- Mit dem nukleären Run-on Assay (engl. nuclear run-on assay) lassen sich RNA-Abschnitte mit transkriptionell aktiver RNA-Polymerase II im Genom identifizieren. Sie beruht darauf, dass gerade entstehende RNA markiert und nachgewiesen wird. Dies geschieht durch Hybridisierung der RNA mit einzelnen Zielsequenzen mittels Dot Blot, oder als Nuclear-run-on-Sequenzierung, basierend auf der Sequenzierung der nächsten Generation, auch auf der Ebene des Transkriptoms.
- In DNA-Microarrays oder -Makroarrays kann die Menge an mRNA einer Vielzahl von Genen aus Zellen einer Kultur/eines Gewebes simultan bestimmt werden. Dazu wird die mRNA isoliert und in cDNA umgeschrieben. Die Detektion erfolgt bei dieser Methode über komplementäre Hybridisierung der markierten cDNA (Radioisotope, Fluoreszenzfarbstoffe) mit den Sonden des DNA-Arrays.
- Mit der Seriellen Analyse der Genexpression (SAGE) und insbesondere SuperSAGE kann die Expression theoretisch aller Gene einer Zelle sehr genau bestimmt werden, indem von jedem Transkript ein kurzes Sequenzstück (das sog. „Tag“ = engl. Etikett) erzeugt wird und möglichst viele dieser Tags sequenziert werden. Vorteil gegenüber Microarrays ist die sehr viel genauere Quantifizierung der Transkripte sowie die Möglichkeit, (v. a. mit SuperSAGE) neue Transkripte (z. B. nichtcodierende Ribonukleinsäuren, wie microRNAs oder antisense-RNAs) zu identifizieren und Organismen mit bisher nicht bekannten Genomen zu untersuchen.
- Beim Differential display (engl. für Differentielle Darstellung, auch DDRT-PCR oder DD-PCR genannt) wird die Änderung der Genexpression auf der Ebene der mRNA zwischen zwei eukaryontischen zellulären Proben verglichen. Da hier zufällig gewählte Primer eingesetzt werden, setzt sie keine Kenntnis über die vorhandenen mRNAs voraus.
- Die Real-Time-quantitative-PCR ist eine Variante der Polymerase-Kettenreaktion (PCR). Durch dem Reaktionsgemisch zugesetzte Farbstoffe oder spezielle Sonden wird die Konzentration des Produktes während der PCR verfolgt. Die zeitliche Änderung der Konzentration ermöglicht Rückschlüsse auf die Ausgangskonzentration der betreffenden Nukleinsäure. Alternativ wird die Digital PCR zur Quantifizierung durchgeführt.
- Mit der „Gesamt-Transkriptom-Shotgun-Sequenzierung“, auch RNA-Seq genannt, die hohe Ansprüche an die bioinformatische Auswertung stellt, versucht man das Transkriptom einer Zelle oder eines Gewebes, also die mengenmäßige Verteilung möglichst aller Transkripte zu bestimmen.
- Ribosomal Profiling, der Nachweis aller RNAs einer Zelle, die zu einem bestimmten Zeitpunkt an Ribosomen gebunden und daher vermutlich translatiert werden, wurde durch das RNA-Seq möglich. Durch die Sequenzierung der Ribosomen-Footprints (Ribo-Seq) ist die Nukleotid-genaue Kartierung möglich.
Protein basiert
Auch auf Proteinebene gibt es eine Vielzahl von Methoden, das Vorkommen einzelner oder einer Vielzahl von Proteinen zu vergleichen.
- Mittels einer klassischen Western-Blot-Analyse lässt sich das Vorkommen einzelner Proteine in verschiedenen biologischen Proben vergleichen. Es werden Proteine hinsichtlich verschiedener Eigenschaften wie Größe, elektrischer Ladung oder Isoelektrischem Punkt aufgetrennt und anschließend mit Antikörpern nachgewiesen. In der Regel wird nur eine überschaubare Anzahl von Genprodukten simultan untersucht.
- Durch die differenzielle Analyse von 2D-Gelen kann die Expression von bis zu 10.000 Proteinen gleichzeitig durchgeführt werden. Dazu werden die Proteinextrakte aus Zellkulturen/Geweben gewonnen, zweidimensional nach dem isoelektrischen Punkt und der Molekülmasse aufgetrennt und mittels Markierung bzw. diverser (Fluoreszenz)färbungstechniken detektiert und quantifiziert. Die ermittelten Intensitäten verschiedener Proben werden miteinander verglichen und somit das Expressionsverhalten über verschiedene Bedingungen verfolgt.
- In Protein-Arrays wird, analog zu DNA-Arrays, die Menge bestimmter Proteine untersucht. Zur Detektion nutzt man die zahlreichen Interaktionen von Proteinen mit anderen Molekülen aus: z. B. Enzym-Substrat-, Antikörper-Antigen- oder Rezeptor-Botenstoff-Interaktion.
- In den letzten Jahren gewinnt die massenspektrometrische Analyse von Proteingemischen immer weiter an Bedeutung. Über die „spectral abundance“ werden Proteinstücke, also Peptide, ähnlich wie bei SAGE die DNA-Fragmente vorhandener RNAs, gezählt und somit deren Häufigkeit quantifiziert. Andere Methoden nutzen die Signalintensität bestimmter Peptide im Massenspektrum als Maß für die Häufigkeit.
Viele Methoden bedienen sich Fluoreszenzfarbstoffen, die an die Sonden (RNA-Sonden, Antikörper etc.) gekoppelt sind und mittels Fluoreszenzspektroskopie oder Fluoreszenzmikroskopie sichtbar gemacht werden. Letztere bietet den Vorteil einer hohen räumlichen Auflösung. Daneben werden auch radioaktiv markierte Sonden eingesetzt, oder solche, die durch gekoppelte Enzyme Chromogene in Farbstoffe umwandeln.
Vergleich mit der Proteomik
Das menschliche Genom enthält ca. 25.000 Gene, welche zusammenarbeiten, um etwa 1.000.000 verschiedene Proteine zu erzeugen. Diese Vielfalt entsteht hauptsächlich durch posttranslationale Modifikationen, so dass ein einzelnes Gen als Vorlage für viele verschiedene Versionen eines Proteins dienen kann. In einem einzelnen Massenspektrometrieexperiment können etwa 2000 Proteine (0,2 % der Gesamtmenge) identifiziert werden. Das Wissen um die einzelnen Proteine, die eine Zelle produziert (Proteomik), ist relevanter als zu wissen, wie viel mRNA von jedem Gen erzeugt wird. Allerdings gibt die Genexpressionsanalyse den bestmöglichen Überblick, den man in einem einzelnen Experiment erhalten kann.
Einschränkungen
Genexpressionsanalysen zeigen Gene auf, deren Expressionsniveau statistisch signifikante Unterschiede zwischen verschiedenen Versuchsbedingungen zeigt. Dies ist aus verschiedenen Gründen meist nur ein kleiner Anteil des gesamten Genoms. Erstens exprimieren verschiedene Zelltypen und Gewebe als direkte Konsequenz ihrer Differenzierung nur einen Teil aller Gene, während die übrigen Gene abgeschaltet sind. Zweitens codieren viele Gene für Proteine, die sich für das Überleben der Zelle in einem bestimmten Konzentrationsbereich befinden müssen, so dass sich ihr Expressionsniveau nicht ändert. Drittens hat eine Zelle neben der Veränderung der mRNA-Menge alternative Mechanismen der Proteinregulation, so dass manche Gene auf gleichbleibendem Niveau exprimiert werden, auch wenn die Menge des Proteins, für das sie codieren, schwankt. Viertens limitieren finanzielle Beschränkungen die Genexpressionsanalyse auf eine kleine Anzahl an Beobachtungen desselben Gens unter identischen Versuchsbedingungen, wodurch die statistische Aussagekraft des Experiments geschwächt wird und kleinere Veränderungen im Expressionsniveau nicht identifiziert werden können. Letztendlich wäre es ein zu großer Aufwand, die biologische Signifikanz eines jeden einzelnen regulierten Gens zu diskutieren, so dass sich Wissenschaftler bei der Diskussion der Ergebnisse einer Genexpressionsanalyse häufig auf eine bestimmte Gruppe von Genen beschränken. Obgleich neuere Microarray-Analysemethoden gewisse Aspekte der Interpretation von Genexpressionsanalyse-Ergebnissen hinsichtlich ihrer biologischen Relevanz automatisieren, bleibt dies nach wie vor eine sehr schwierige Herausforderung. Da die Listen publizierter Gene aus Genexpressionsanalysen meist relativ kurz sind, ist ein Vergleich des Grades an Übereinstimmung mit Ergebnissen eines anderen Labors eingeschränkt. Das Einstellen der Ergebnisse von Genexpressionsanalysen in eine öffentliche Datenbank (Microarray database) ermöglicht es Wissenschaftlern, Expressionsmuster über die in Publikationen enthaltenen Informationen hinaus zu vergleichen und ggf. Übereinstimmungen mit ihren eigenen Daten zu finden.
Validierung von Hochdurchsatz-Methoden
Sowohl DNA-Microarrays als auch qPCR nutzen die bevorzugte Bindung komplementärer Nukleinsäuresequenzen („base pairing“), und beide Methoden kommen, häufig aufeinander folgend, beim Erstellen von Genexpressionsprofilen zum Einsatz. Während DNA-Microarrays eine hohe Durchsatzrate haben, fehlt ihnen die hohe quantitative Genauigkeit der qPCR. Allerdings kann man in der gleichen Zeit, die man benötigt, um die Expression einiger Dutzend Gene mit qPCR zu bestimmen, das gesamte Genom mittels DNA-Microarrays untersuchen. Aus diesem Grund ist es häufig sinnvoll, zunächst eine semiquantitative DNA-Microarray-Analyse zur Identifizierung von Kandidatengenen durchzuführen, welche dann im Anschluss mittels qPCR validiert und genauer quantifiziert werden können. Zusätzliche Experimente, wie etwa Western Blots der Proteine differentiell exprimierter Gene, können dazu beitragen, die Ergebnisse der Genexpressionsanalyse zu untermauern, da mRNA-Konzentrationen nicht unbedingt mit der Menge an exprimiertem Protein korrelieren.
Statistische Analyse
Die Analyse von Microarray-Daten hat sich zu einem intensiven Forschungsgebiet entwickelt. Die früher übliche Praxis, anzugeben, dass eine Gruppe von Genen um den Faktor 2 hoch- oder runterreguliert ist, entbehrt einer soliden statistischen Grundlage. Mit den für Microarrays typischen 5 oder weniger Wiederholungen in jeder Gruppe kann bereits ein einzelner Ausreißer einen scheinbar mehr als 2-fachen Unterschied hervorrufen. Darüber hinaus fußt die willkürliche Festlegung, dass Abweichungen ab einem Faktor von 2 signifikant sind, nicht auf einer biologischen Grundlage, da hierdurch viele Gene mit offensichtlicher biologischer Signifikanz (aber zu geringer Schwankung in der Expression) ausgeschlossen werden. Anstatt also differentiell exprimierte Gene mittels eines Schwellenwerts für eine x-fache Veränderung zu identifizieren, können verschiedene statistische Tests oder Omnibus-Tests wie etwa ANOVA verwendet werden. Derartige Tests berücksichtigen sowohl x-fache Veränderung als auch die Variabilität, um einen p-Wert zu erzeugen, welcher ein Maß dafür ist, wie häufig diese Daten rein zufällig beobachtet werden könnten. Die Anwendung dieser Tests auf Microarray-Daten wird durch die große Anzahl verschiedenster Vergleichsmöglichkeiten zwischen den einzelnen Genen erschwert. So wird beispielsweise ein p-Wert von 0.05 für gewöhnlich als Zeichen dafür gewertet, dass die Daten signifikant sind, da dieser Wert eine nur 5%ige Wahrscheinlichkeit dafür angibt, dass diese Daten rein zufällig beobachtet werden können. Schaut man allerdings 10.000 Gene auf einem Microarray an, so bedeutet p<0.05, dass 500 dieser Gene fälschlicherweise als signifikant hoch oder runterreguliert identifiziert werden, obgleich kein wirklicher Unterschied zwischen den verschiedenen Versuchsbedingungen existiert. Eine offensichtliche Lösung dieses Problems besteht darin, nur solche Gene zu berücksichtigen, die stringenteren p-Wert-Kriterien entsprechen, z. B. durch Verwendung einer Bonferroni-Korrektur oder durch Anpassung der p-Werte an die Anzahl parallel durchgeführter Versuche unter Berücksichtigung der Falsch-Positiv-Rate. Unglücklicherweise können derartige Ansätze die Zahl signifikanter Gene auf Null reduzieren, sogar wenn einige dieser Gene tatsächlich differentiell exprimiert werden. Gängige Methoden der Statistik wie die Rangprodukt-Methode versuchen ein Gleichgewicht zu erreichen zwischen der Falscherkennung von Genen aufgrund zufälliger Veränderungen (false positives) auf der einen Seite und der Nichterkennung differentiell exprimierter Gene (false negatives) auf der anderen Seite. Zu den häufig zitierten Methoden zählt die Signifikanz-Analyse von Microarrays (SAM) sowie zahlreiche weitere Methoden, erhältlich in Software-Paketen von Bioconductor oder anderen Bioinformatikfirmen. Die Wahl eines anderen Tests liefert für gewöhnlich eine andere Liste signifikanter Gene, da jeder Test einen spezifischen Satz von Annahmen verwendet und einen unterschiedlichen Schwerpunkt auf bestimmte Eigenschaften der Daten legt. Viele Tests basieren auf der Annahme einer Normalverteilung der Daten, da dies häufig ein vernünftiger Startpunkt ist und Ergebnisse mit scheinbar höherer Signifikanz liefert. Manche Tests berücksichtigen die gemeinsame Verteilung aller Gen-Beobachtungen, um die allgemeine Variabilität der Messungen abzuschätzen, während andere jedes einzelne Gen für sich betrachten. Viele moderne Microarray-Analysetechniken involvieren die Bootstrap-Technik, maschinelles Lernen (z. B. mit Random Forest) oder Monte-Carlo-Methoden. Je höher die Anzahl von Wiederholungsversuchen in einem Microarray-Experiment ist, desto ähnlicher werden die Ergebnisse verschiedener statistischer Analysemethoden. Im Gegensatz dazu erscheinen Microarrays mit einer geringen Übereinstimmung der Ergebnisse verschiedener Methoden der statistischen Datenanalyse weniger zuverlässig. Das MAQC Project gibt Wissenschaftlern Empfehlungen für die Wahl von Standardmethoden, mit Hilfe derer Experimente verschiedener Labore besser miteinander übereinstimmen.
Verwendung zur Entwicklung und Überprüfung von Hypothesen
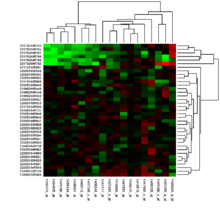
Manchmal hat ein Wissenschaftler bereits eine Hypothese und führt eine Genexpressionsanalyse durch, um diese Hypothese zu überprüfen. Mit anderen Worten, der Wissenschaftler macht eine spezifische Vorhersage hinsichtlich der zu erwartenden Expressionsniveaus, welche sich als richtig oder falsch herausstellen kann. Häufig werden Genexpressionsanalysen durchgeführt, bevor bekannt ist, wie sich die jeweiligen Versuchsbedingungen auf die Expression bestimmter Gene auswirken. Das heißt, es gibt zunächst keine überprüfbare Hypothese, doch die Genexpressionsanalyse kann dazu beitragen, Kandidatengene für zukünftige Experimente zu identifizieren. Die meisten der frühen sowie viele der heutigen Genexpressionsanalysen haben diese Form, welche als Klassen-Feststellung (class discovery) bezeichnet wird. Ein beliebter Ansatz der Klassen-Feststellung beinhaltet die Einteilung ähnlicher Gene oder Proben in Clustern mit Hilfe des k-Means-Algorithmus oder hierarchischer Clusteranalyse. Die Abbildung (s. o.) zeigt das Ergebnis eines zweidimensionalen Clusters, in welchem ähnliche Proben (Reihen) und ähnliche Gene (Spalten) so organisiert sind, dass sie dicht beieinander liegen. Die einfachste Form der Klassen-Feststellung ist eine Auflistung aller Gene, die sich unter verschiedenen experimentellen Bedingungen um mehr als einen bestimmten Wert verändert haben. Die Klassen-Vorhersage ist schwieriger als die Klassen-Feststellung, doch sie erlaubt die Beantwortung von Fragen mit direkter klinischer Relevanz, wie etwa die Frage, wie hoch die Wahrscheinlichkeit ist, dass ein Patient mit einem bestimmten Profil auf ein zu untersuchendes Medikament ansprechen wird. Hierfür sind viele Beispiele von Profilen von Patienten, die auf das Medikament angesprochen bzw. nicht angesprochen haben, nötig, sowie Methoden der Vergleichsprüfung (Kreuzvalidierungsverfahren) zur Unterscheidung zwischen diesen Profilen.
Aufgrund der hohen Zahl untersuchter Gene, können bei nicht Beachtung der Alphafehler-Kumulierung Genexpressionen fälschlicherweise als signifikant festgestellt werden.
Gen-Annotation
Mit Hilfe statistischer Methoden können Gene, deren Produkte sich unter experimentellen Bedingungen verändern, zuverlässig identifiziert werden. Für eine sinnvolle Interpretation von Expressionsprofilen ist es unabdingbar zu wissen, welches Protein von welchem Gen codiert wird und welche Funktion es hat. Dieser Prozess wird Gen-Annotation genannt. Manche Annotationen sind zuverlässiger als andere, und mitunter fehlen sie vollständig. Gen-Annotations-Datenbanken ändern sich ständig, und verschiedene Datenbanken verwenden unterschiedliche Bezeichnungen für dasselbe Protein, worin sich ein Wandel im Verstehen seiner Funktion widerspiegelt. Die Verwendung einer standardisierten Gen-Nomenklatur vermeidet das Problem verschiedener Namensgebung, doch die exakte Zuordnung von Transkripten zu Genen bleibt eine wichtige Herausforderung.
Klassifizierung regulierter Gene
Der nächste Schritt nach der Identifizierung einer Gruppe differenziell regulierter Gene ist die Suche nach Mustern innerhalb dieser Gruppe. Haben die Proteine, für die diese Gene codieren, ähnliche Funktionen? Sind sie sich chemisch ähnlich? Sind sie in ähnlichen Zellkompartimenten lokalisiert? Die Analyse der Gen-Ontologie bietet eine gängige Möglichkeit, diese Beziehungen zu definieren. Gen-Ontologie fängt mit einer sehr weit gefassten Oberkategorie an, z. B. „Stoffwechselprozess“, und unterteilt diese dann in kleinere Subkategorien wie etwa „Kohlenhydratstoffwechsel“, welche dann ihrerseits noch einmal in spezifischere Untergruppen wie „Phosphorylierung von Inositol und Derivaten“ aufgeteilt werden können. Neben ihrer biologischen Funktion, chemischen Eigenschaften und zellulären Lokalisation haben Gene noch andere Eigenschaften. So kann man Gene beispielsweise anhand ihrer Verwandtschaft zu anderen Genen, ihrem Zusammenhang mit Erkrankungen oder aber ihrem Zusammenspiel mit Medikamenten oder Toxinen in Gruppen einordnen. Die Datenbank molekularer Signaturen (Molecular Signature Database) und die Vergleichende Toxikogenomische Datenbank (Comparative Toxicogenomics Database) bieten die Möglichkeit, Gene auf die unterschiedlichsten Arten zu kategorisieren.
Mustererkennung zwischen regulierten Genen

Wenn regulierte Gene danach geordnet werden, was sie sind und was sie machen, können wichtige Beziehungen zwischen verschiedenen Genen zu Tage treten. So könnte man beispielsweise einen Hinweis darauf erhalten, dass ein bestimmtes Gen ein Protein codiert, das ein Enzym erzeugt, das dann seinerseits ein Protein aktiviert, welches dann ein zweites Gen auf unserer Liste reguliert. Dieses zweite Gen könnte ein Transkriptionsfaktor sein, der wiederum ein anderes unserer Kandidatengene reguliert. Durch Beobachtung dieser Querverbindungen können wir vermuten, dass es sich hierbei um mehr als zufällige Assoziationen handelt und dass all diese Gene auf unserer Liste sind, da sie Teil eines zugrunde liegenden biologischen Prozesses sind. Andererseits könnten natürlich auch voneinander unabhängige, völlig zufällig ausgewählte Gene den Anschein erwecken, dass sie Teil eines gemeinsamen Prozesses sind, obgleich dies nicht der Fall ist.
Kausalitätsbeziehungen
Mit Hilfe einfacher statistischer Mittel kann man abschätzen, ob Assoziationen zwischen verschiedenen Genen einer Liste größer sind, als es rein zufällig der Fall wäre. Diese einfachen Statistiken sind interessant, auch wenn sie eine sehr starke Vereinfachung der tatsächlichen Situation darstellen.
Während Genprofile an sich, aufgrund der im Beispiel unten genannten Vorbehalte, keinen kausalen Zusammenhang zwischen experimentellen Bedingungen und biologischen Auswirkungen beweisen können, so bieten sie doch einzigartige biologische Einsichten in Zusammenhänge, die nur sehr schwer auf anderem Weg zu erlangen wären. Diese Zusammenhänge zwischen verschiedenen Genen über die von ihnen exprimierten Regulatorproteine werden mit Genregulationsnetzwerken graphisch abgebildet. Derartige Netzwerkmodelle werden mit bioinformatischen Methoden im Ergebnis der Genexpressionsanalyse in Verbindung mit Vorwissen (Prior knowlege) aus molekularbiologischen Datenbanken identifiziert. Diese datenbasierte Netzwerkmodellierung wird Netzwerkinferenz genannt.
Beispiel
Angenommen, ein Experiment umfasst 10.000 Gene, von denen nur 50 (0,5 %) bekannterweise eine Rolle bei der Cholesterinsynthese spielen. Das Experiment identifiziert 200 regulierte Gene. 40 (20 %) dieser Gene befinden sich auch auf einer Liste von Cholesterin-Genen. Basierend auf der Gesamthäufigkeit von Cholesterin-Genen (0,5 %) würde man im Schnitt nur 1 Cholesterin-Gen pro 200 regulierten Genen erwarten. Dies ist lediglich ein Durchschnittswert, d. h. mitunter kann man auch mehr als 1 Gen von 200 erwarten. Die Frage ist nun, wie häufig sähe man 40 Gene statt 1 Gen aus reinem Zufall? Gemäß der hypergeometrischen Verteilung bräuchte man etwa Versuche (eine 10 mit 56 Nullen), bevor man aus einem Pool von 10.000 Genen rein zufällig 200 Gene auswählen würde, von denen dann 39 oder mehr Cholesterin-Gene sind. Ob man nun erkennt, wie verschwindend gering die Wahrscheinlichkeit ist, dass es sich hierbei um eine Zufallsbeobachtung handelt, oder nicht, man käme in jedem Fall zu dem Schluss, dass die Liste regulierter Gene mit Cholesterin-assoziierten Genen angereichert ist. Darüber hinaus könnte man die Hypothese aufstellen, dass die experimentellen Bedingungen einen Einfluss auf die Cholesterin-Regulation haben, da diese selektiv jene Gene regulieren, die mit Cholesterin assoziiert sind. Obgleich dies wahr sein könnte, so gibt es doch eine Vielzahl von Gründen, weshalb es unangebracht ist, hier voreilige Schlussfolgerungen zu ziehen. Zum einen hat die Genregulation nicht zwingend einen direkten Einfluss auf die Proteinregulation. Sogar wenn die Proteine, für die diese Gene codieren, nichts anderes tun, als Cholesterin zu produzieren, so sagt eine Veränderung ihrer mRNA-Konzentration noch nichts über Änderungen auf der Proteinebene aus. Es ist gut möglich, dass die Menge dieser Cholesterin-assoziierten Gene unter den Versuchsbedingungen konstant bleibt. Zum anderen wäre es möglich, dass selbst wenn sich die Proteinmengen verändern, es dennoch stets genug Protein gibt, um die Cholesterinsynthese mit maximaler Geschwindigkeit aufrechtzuerhalten, da nicht diese Proteine, sondern ein weiteres Protein, welches nicht auf der Liste ist, den geschwindigkeitsbestimmenden Schritt und limitierenden Faktor der Cholesterinsynthese darstellt. Zudem haben Proteine typischerweise viele verschiedene Funktionen, so dass diese Gene nicht aufgrund ihrer gemeinsamen Assoziation mit der Cholesterinsynthese reguliert werden, sondern aufgrund einer gemeinsamen Funktion in einem völlig unabhängigen Prozess.

Verwendung von Mustern zur Erkennung regulierter Gene
Wie oben beschrieben, kann man zuerst Gene mit signifikant veränderter Expression identifizieren und anschließend Zusammenhänge zwischen Gruppen verschiedener signifikanter Gene finden durch Vergleich einer Liste signifikanter Gene mit einer Gruppe von Genen mit bekannter Assoziation. Man kann das Problem auch andersherum lösen, hier ein Beispiel: 40 Gene stehen im Zusammenhang mit einem bekannten Prozess, z. B. einer Veranlagung für Diabetes. Man nehme zwei Gruppen von Expressionsprofilen, eine mit Mäusen, die kohlenhydratreich gefüttert wurden, und die andere mit Mäusen auf einer kohlenhydratarmen Diät. Beim Vergleich dieser beiden Gruppen von Expressionsprofilen beobachtet man, dass alle 40 Diabetes-Gene in der Gruppe mit der kohlenhydratreichen Ernährung in stärkerem Maße exprimiert werden, als in der kohlenhydratarmen Gruppe. Unabhängig davon, ob irgendeines dieser Gene auf einer Liste signifikant verändert exprimierter Gene gelandet wäre oder nicht, kann die Tatsache, dass alle 40 Gene herauf und keines herunter reguliert wurde, kaum das Ergebnis reinen Zufalls sein. Es wäre, wie beim Werfen von Münzen 40-mal hintereinander „Kopf“ zu werfen, was einer Wahrscheinlichkeit von eins zu einer Billion entspricht. Eine Gruppe von Genen, deren kombiniertes Expressionsmuster den Versuchsbedingungen eine einzigartige Charakteristik verleiht, stellt die Gensignatur dieser Bedingungen für eine bestimmte Zellart dar. Idealerweise kann eine solche Gensignatur dazu verwendet werden, eine Gruppe von Patienten zu identifizieren, die sich in einem bestimmten Krankheitsstadium befinden, wodurch die Auswahl der adäquaten Behandlungsmethode erleichtert wird. Gene Set Enrichment Analysis (GSEA) basiert auf dieser Art von Logik unter Verwendung anspruchsvollerer statistischer Methoden, da Gene häufig ein komplexeres Verhalten zeigen, als einfach als Gruppe hoch- oder herunterreguliert zu sein. Zudem ist das Ausmaß der Hoch- oder Runterregulation entscheidend und nicht nur die Richtung. In jedem Fall messen derartige statistische Methoden, inwieweit sich das Verhalten kleiner Gruppen von Genen von anderen Genen unterscheidet, die nicht zu dieser Gruppe gehören. GSEA verwendet eine Statistik im Stil von Kolmogorov/Smirnov, um zu bestimmen, ob irgendwelche zuvor definierten Gengruppen ein ungewöhnliches Verhalten im aktuellen Expressionsprofil an den Tag legen. Dies führt zu der Herausforderung des Testens mehrerer Hypothesen, doch es gibt angemessene Methoden, um dieses Problem anzugehen.
Schlussfolgerungen
Die Genexpressionsanalyse liefert neue Informationen darüber, wie sich Gene unter verschiedensten Bedingungen verhalten. Im Großen und Ganzen produzieren Microarray-Techniken zuverlässige Expressionsprofile. Basierend auf diesen Daten können neue biologische Hypothesen aufgestellt oder bestehende Hypothesen überprüft werden. Allerdings führen Umfang und Komplexität dieser Experimente häufig zu einer Vielzahl verschiedener Interpretationsmöglichkeiten. In vielen Fällen erfordert die Datenanalyse von Expressionsprofilen wesentlich mehr Zeit und Mühe als das ursprüngliche Experiment zur Generierung der Daten. Viele Wissenschaftler verwenden mehrere statistische Methoden und orientierende Datenanalyse und konsultieren Biostatistiker oder andere Experten im Gebiet der Microarray Technik, bevor sie die Ergebnisse von Genexpressionsanalysen veröffentlichen. Ein guter Versuchsaufbau, eine adäquate Zahl biologischer Replikate sowie Wiederholungsexperimente spielen eine Schlüsselrolle bei der Durchführung erfolgreicher Genexpressionsanalysen.
Anwendungen
Bei den klassischen Verfahren zur Krankheitsbestimmung wird der Phänotyp (die äußerlich diagnostizierbare Auswirkung molekularer Ereignisse) nachgewiesen. Mit den modernen Methoden können die spezifischen molekularen Veränderungen detektiert werden, die einer Krankheit ursächlich zugrunde liegen. Somit führt der Nachweis krankheitsrelevanter Moleküle nicht nur zu einem besseren Verständnis von Krankheitsprozessen, sondern kann auch Diagnosen zu einem früheren Zeitpunkt im Krankheitsverlauf ermöglichen.