
| Klassifikation nach ICD-10 | |
|---|---|
| G71.0 | Muskeldystrophie - Becken- oder Schultergürtelform |
| ICD-10 online (WHO-Version 2019) | |
Der Begriff Gliedergürteldystrophie (engl. limb-girdle muscular dystrophy, LGMD) bezeichnet eine Gruppe erblicher Muskelerkrankungen (Myopathien), deren gemeinsames Merkmal Lähmungen der Muskulatur des Schulter- und Beckengürtels sind. Schulter- und Beckengürtel werden in der Medizin zusammen als Gliedergürtel bezeichnet. Neben Muskellähmungen erfüllen die Gliedergürteldystrophien die Kriterien der Muskeldystrophie. Eine Muskeldystrophie ist gekennzeichnet durch Umbauprozesse des Muskelgewebes. Die Erkrankungen dieser Gruppe sind genetisch und klinisch heterogen. Sie werden durch unterschiedliche Genmutationen verursacht und die gleichen Genmutationen können ein sehr variables klinisches Bild hervorrufen. Der Beginn der Erkrankung reicht vom Kleinkindes- bis in das hohe Erwachsenenalter und es sind sowohl milde als auch schwere Verläufe möglich. Gliedergürteldystrophien sind seltene Erkrankungen. Schätzungen der Krankheitshäufigkeit (Prävalenz) aller Gliedergürteldystrophien reichen von 1:14.500 bis 1:123.000.
Ursachen
Die Gliedergürteldystrophien werden durch Gendefekte auf verschiedenen Chromosomen verursacht. Diese führen zu struktureller Abweichung, zum Mangel oder zum völligen Fehlen von verschiedenen Muskelproteinen, was zu einer chronischen Schädigung der häufig beanspruchten Muskelfasern führt. Leitsymptom ist die zunehmende Schwäche und der sichtbare Schwund der betroffenen Muskelpartien.
Einteilung
Die heute übliche Einteilung basiert auf der Klassifikation der molekulargenetisch nachweisbaren Genmutationen bzw. der veränderten Proteine. Nach dem Erbgang werden 2 Gruppen unterschieden: die autosomal-dominant vererbten Gliedergürteldystrophien, LGMD1, und die autosomal-rezessiv vererbten Gliedergürteldystrophien, LGMD2.
Autosomal-dominante Formen der Gliedergürteldystrophien
Die autosomal-dominant vererbten Gliedergürteldystrophien sind relativ selten und machen etwa 10 % aller Gliedergürteldystrophien aus. Die Erkrankungen manifestieren sich meist im Erwachsenenalter (adult-onset) und der klinische Verlauf ist im Vergleich zu den autosomal-rezessiv-vererbten Gliedergürteldystrophien häufig weniger schwer.
| Typ | Genort | Gen | Genprodukt | Manifestationsalter | Leitsymptome | Prognose |
|---|---|---|---|---|---|---|
| LGMD1A | 5q22–q34 | MYOT | Myotilin | 18–35 | Schwäche hüftnaher Beinmuskeln, Jahre später sind Armmuskeln betroffen; Schwäche von Gesichts- und Schlundmuskeln bei 20 %, Sprechstörungen (Dysarthrie) bei 25 % | Rollstuhlabhängigkeit etwa 20 Jahre nach Beginn, meist normale Lebenserwartung |
| LGMD1B | 1q22 | LMNA | Lamin-A/C | 4–30 | Schwächen in Hüft- und hüftnahen Oberschenkelmuskeln, in zwei Drittel der Fälle Reizüberleitungsstörungen des Herzen und möglicherweise Herzmuskelschwäche mit Erweiterung des Herzen (dilatative Kardiomyopathie) | Lebenslange Gehfähigkeit, jedoch Risiko des plötzlichen Herztods um 50–60 Jahre |
| LGMD1C | 3p25.3 | CAV3 | Caveolin-3 | 2–20–70 | Stammnahe Muskelschwäche, Wadenkrämpfe, Wadenpseudohypertrophie | wahrscheinlich lebenslange Gehfähigkeit und normale Lebenserwartung |
| LGMD1D | 6q23 | DES | Desmin | 20–25, selten unter 20 | Herzrhythmusstörungen und Kardiomyopathie, später zusätzliche stammnahe Muskelschwächen | Plötzlicher Tod durch AV-Block, ventrikuläre Tachykardie oder Kardiomyopathie möglich |
| LGMD1E | 7q | nicht bekannt | nicht bekannt | 10–30 | Hüftnahe Beinmuskelschwäche | |
| LGMD1F | 7q32.1 | TNPO3 | Transportin 3 | unter 1–58 | Beckengürtel-, Schultergürtelmuskelschwäche | |
| LGMD1G | 4q21 | HNRNPDL | D-ähnliches heterogenes nukleäres Ribonukleoprotein | 30–47 | Becken- und Schultergürtelmuskelschwäche, Beugekontrakturen der Zehen und Finger | |
| LGMD1H | 3p25.1-p23 | nicht bekannt | nicht bekannt |
Autosomal-rezessive Formen der Gliedergürteldystrophien
| Typ | Genort | Gen | Genprodukt | Häufigkeit | Manifestationsalter | Leitsymptome | Prognose |
|---|---|---|---|---|---|---|---|
| LGMD2A | 15q15.1 | CAPN3 | Calpain-3 | Etwa 10 % aller LGMD2, Gründermutationen: Amische, La Réunion, Basken, Türken | 3–10–30 | Schwäche der Hüft- und Oberschenkelmuskeln, auch Rumpfmuskulatur betroffen, Schultermuskulatur oft 2–5 Jahre später, häufig auch Kontrakturen der Wirbelsäule, Fußgelenke, Ellenbogen und Hände | rascher oder langsamer Verlauf, entsprechend Tod um 20. Lebensjahr möglich, sonst in der 8. Lebensdekade, früher Beginn bedeutet nicht automatisch rasches Fortschreiten |
| LGMD2B | 2p13.2 | DYSF | Dysferlin | Etwa 5 % aller LGMD2 | 13–22–35 | Schwäche vor allem der hinteren Hüft- und hüftnahen Oberschenkelmuskeln, 25 % Wadenpseudohypertrophie, 2–10 Jahre später Schultermuskelschwäche | rascher oder langsamer Verlauf, in etwa 30 % Gehfähigkeit bis 26–54 Jahren, sonst bis 8. Dekade |
| LGMD2C | 13q12.12 | SGCG | Gamma-Sarkoglykan | 3–12 | Muskelschwäche im Beckengürtel früher als im Schultergürtel, frühe Wadenpseudohypertrophie, später möglicherweise Gesichtsmuskeln betroffen | Gehfähigkeit in 25 % 10–15 Jahre, 50 % 15–20 Jahre, 25 % über 20 Jahre, Tod im zweiten Lebensjahrzehnt möglich | |
| LGMD2D | 17q21.33 | SGCA | Alpha-Sarkoglykan | 1–16, später | Zunehmende Muskelschwächen im Beckengürtel und Oberschenkel mit verspäteten Gehbeginn oder unsicherem Gang ähnlich wie bei Muskeldystrophie Typ Duchenne | rascher oder langsamer Verlauf, Gehfähigkeit bis 16–30 Jahre oder später, vorzeitiger Tod bei raschem Verlauf möglich | |
| LGMD2E | 4q12 | SGCB | Beta-Sarkoglykan | 3–12 | Beckengürtel eher betroffen als Schultergürtel, Wadenpseudohypertrophie | rascher oder langsamer Verlauf, Gehfähigkeit 9–14 Jahre oder bis 38 Jahre, Tod im 2.–3. Lebensjahrzehnt möglich | |
| LGMD2F | 5q33–q34 | SGCD | Delta-Sarkoglykan | 4–10 | Zunehmende Muskelschwächen im Beckengürtel und Oberschenkel mit verspäteten Gehbeginn oder unsicherem Gang ähnlich wie bei Muskeldystrophie Typ Duchenne, Gesichtsmuskeln möglicherweise betroffen, Wadenhypertrophie | rasches Fortschreiten mit Gehfähigkeit 9–16 Jahre, Tod am Ende des ersten oder im zweiten Lebensjahrzehnt möglich | |
| LGMD2G | 7q12 | TCAP | Telethonin | Etwa 3 % aller LGMD2 | 9–15 | Neben Hüft- und Oberschenkelmuskelschwäche auch Unterschenkelmuskeln und Arm/Schultermuskeln betroffen | lange Gehfähigkeit bis 18–25 Jahre nach Beginn |
| LGMD2H | 9q33.1 | TRIM32 | E3-Ubiquitin-Protein-Ligase TRIM32 | 1.–3. Dekade | Beckengürtel eher betroffen als Schultergürtel, möglicherweise Nacken- oder Rückenschmerzen | Gehschwierigkeiten mit 37–46 Jahren, Rollstuhlabhängigkeit im siebten Lebensjahrzehnt | |
| LGMD2I | 19q13.32 | FKRP | Fukutin-assoziiertes Protein (FKRP) | Etwa 6 % aller LGMD2 | 1.–4 Dekade | Zunächst Schwäche der Hüft- und Oberschenkelmuskeln, später der Schulter- und Oberarmmuskeln, Wadenpseudohypertrophie, Muskelschmerzen bei Belastung, Muskelfaserzerfall (Rhabdomyolyse) | sehr unterschiedlicher Verlauf, Gehfähigkeit meist bis 30 Jahre |
| LGMD2J | 2q31.2 | TTN | Titin | 1.–3. Dekade | Muskelschwäche und -atrophie der Hüft- und hüftnahen Oberschenkelmuskeln | Gehverlust im 3.–5. Lebensjahrzehnt | |
| LGMD2K | 9q34.13 | POMT1 | Protein-O-Mannosyltransferase 1 | ||||
| LGMD2L | 11p14.3 | ANO5 | Anoctamin-5 | Etwa 25 % der LGMD2 im Vereinigten Königreich, Gründermutationen: Nordeuropäer | |||
| LGMD2M | 9q31.2 | FKTN | Fukutin | 6 Patienten | |||
| LGMD2N | 14q24.3 | POMT2 | Protein-O-Mannosyltransferase 2 | 2 Patienten | |||
| LGMD2O | 1p34.1 | POMGNT1 | Protein-O-Mannose-beta-1,2-N-Acetylglucosaminyltransferase 1 | 2 unabhängige Patienten (1 irisches Mädchen, 1 belgischer Junge) | |||
| LGMD2P | 3p21.31 | DAG1 | Dystroglycan | 1 türkische Patientin, homozygote Mutation | frühe Kindheit | Gliedergürteldystrophie mit mentaler Retardierung | Mit 16 Jahren Laufen weniger Meter, IQ von 50 |
| LGMD2Q | 8q24.3 | PLEC1 | Plectin | 4 Mitglieder einer türkischen Familie | |||
| LGMD2R | 2q35 | DES | Desmin | 2 türkische Sippen | |||
| LGMD2S | 4q35.1 | TRAPPC11 | TRAPPC11 | 3 syrische Familien, 5 Hutterer | |||
| LGMD2T | 3p21.31 | GMPPB | Beta-GDP-Mannose-Phosphorylase | 3 unabhängige Patienten | |||
| LGMD2U | 7p21.2 | ISPD | Isoprenoid synthase domain-containing protein | ||||
| LGMD2V | 17q25.3 | GAA | Lysosomale Alpha-Glucosidase | ||||
| LGMD2W | 3p21.31 | LIMS2 | LIM and senescent cell antigen-like-containing domain protein 2 | Kindesalter | Gliedergürteldystrophie mit Pseudohypertrophie der Waden und Makroglossie, eingeschränkte linksventrikuläre Funktion ab der 3. Dekade | ||
| LGMD2X | 6q21 | BVES | Blood vessel epicardial substance (Popeye protein 1) | Erwachsenenalter |
Diagnose
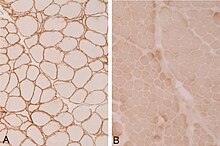
In der Ableitung der elektrischen Muskelaktivität (Elektromyografie, EMG) finden sich unspezifische Hinweise auf eine chronische Muskelschädigung (Myopathie). Durch bildgebende Verfahren wie die Computertomografie (CT) oder die Magnetresonanztomografie (MRT) lassen sich besonders befallene Muskelgruppen darstellen. Labordiagnostisch ist häufig eine deutlich erhöhte Creatinkinase auffällig. Bei entsprechendem Verdacht durch klinische Symptome helfen meist immunhistochemische oder molekulargenetische Untersuchungen einer Muskelbiopsie, die Diagnose zu sichern.
Therapie
Eine etablierte medikamentöse Therapie ist bei keiner der Gliedergürteldystrophien bekannt. Die symptomatische Behandlung konzentriert sich auf krankengymnastische Maßnahmen zum Erhalt der Muskelkraft, zur Schulung von Alltagsbewegungen und zur Vorbeugung gegen Kontrakturen. Ein maximales Krafttraining gilt jedoch als ungünstig. Wichtig ist auch eine sachgerechte Hilfsmittelversorgung in Form von Schienen (Orthesen), Gehstock, Rollator oder einem Rollstuhl. Bei Fehlstellungen der Füße oder der Wirbelsäule kommen insbesondere zur Wiederherstellung der Gehfähigkeit auch operative Behandlungsmaßnahmen in Frage. Bei einer auftretenden Beeinträchtigung der Sprechmotorik (Dysarthrie) ist eine logopädische Behandlung notwendig.
Nach Operationen oder anderen Umständen einer eingeschränkten Mobilität ist eine frühe Mobilisierung wichtig, da eine längere Immobilisierung von Patienten mit Gliedergürteldystrophie nicht selten dazu führt, dass die Gehfähigkeit verloren geht. Die Physiotherapie sollte bereits am Tag nach Operationen begonnen und konsequent durchgeführt werden.
Bei Herzbeteiligung ist unter Umständen eine Therapie von Reizleitungsstörungen und anderen Erkrankungen des Herzens möglich.
Medizingeschichte
Meist wird der Begriff der Gliedergürteldystrophien auf eine Arbeit von John Nicholas Walton und Frederick John Natrass aus dem Jahr 1954 zurückgeführt. Erste Beschreibungen von Gliedergürteldystrophien gehen bereits auf Arbeiten von Ernst von Leyden und Paul Julius Möbius in den Jahren 1876 beziehungsweise 1879 zurück.
Literatur
- V. Nigro, M. Savarese: Genetic basis of limb-girdle muscular dystrophies: the 2014 update. In: Acta myologica: myopathies and cardiomyopathies: official journal of the Mediterranean Society of Myology / edited by the Gaetano Conte Academy for the study of striated muscle diseases. Band 33, Nummer 1, Mai 2014, S. 1–12, ISSN 1128-2460. PMID 24843229. PMC 4021627 (freier Volltext).
- E. Pegoraro, E. P. Hoffman: Limb-Girdle Muscular Dystrophy Overview. GeneReviews [Internet]. University of Washington, Seattle, 8. Juni 2000, [update vom 30. August 2012]. PMID 20301582.
- L. Broglio, M. Tentorio u. a.: Limb-girdle muscular dystrophy-associated protein diseases. In: The neurologist. Band 16, Nummer 6, November 2010, S. 340–352, ISSN 1074-7931. doi:10.1097/NRL.0b013e3181d35b39. PMID 21150381. (Review).
- C. T. Rocha, E. P. Hoffman: Limb-girdle and congenital muscular dystrophies: current diagnostics, management, and emerging technologies. In: Current neurology and neuroscience reports. Band 10, Nummer 4, Juli 2010, S. 267–276, ISSN 1534-6293. doi:10.1007/s11910-010-0119-1. PMID 20467841. (Review).
- K. Bushby: Diagnosis and management of the limb girdle muscular dystrophies. In: Practical neurology. Band 9, Nummer 6, Dezember 2009, S. 314–323, ISSN 1474-7766. doi:10.1136/jnnp.2009.193938. PMID 19923111. (Review).
- A. Ferbert, W. Kress: Klinik und Genetik der Gliedergürteldystrophien. In: Medizinische Genetik. Volume 21, Number 3, 2009, S. 332–336, doi:10.1007/s11825-009-0171-x
- M. Guglieri, K. Bushby: How to go about diagnosing and managing the limb-girdle muscular dystrophies. In: Neurology India. Band 56, Nummer 3, 2008 Jul.–Sep., S. 271–280, ISSN 0028-3886. PMID 18974553. (Review).
- M. Guglieri, V. Straub u. a.: Limb-girdle muscular dystrophies. In: Current Opinion in Neurology. Band 21, Nummer 5, Oktober 2008, S. 576–584, ISSN 1350-7540. doi:10.1097/WCO.0b013e32830efdc2. PMID 18769252. (Review).
- F. Norwood, M. de Visser u. a.: EFNS guideline on diagnosis and management of limb girdle muscular dystrophies. In: European journal of neurology. Band 14, Nummer 12, Dezember 2007, S. 1305–1312, ISSN 1468-1331. doi:10.1111/j.1468-1331.2007.01979.x. PMID 18028188. (Review).
- V. Straub, K. Bushby: The childhood limb-girdle muscular dystrophies. In: Seminars in pediatric neurology. Band 13, Nummer 2, Juni 2006, S. 104–114, ISSN 1071-9091. doi:10.1016/j.spen.2006.06.006. PMID 17027860. (Review).
- D. Fischer: Klinische und bildgebende Differenzialdiagnose von Gliedergürteldystrophien. In: Klin Neurophysiol. Band 37, Nr. 3, 2006, S. 180–188, doi:10.1055/s-2006-940133.
- J. Finsterer: Klinik und Genetik der Gliedergürteldystrophien. In: Der Nervenarzt. 2004, Volume 75, Number 12, S. 1153–1166, doi:10.1007/s00115-004-1769-5.
- J. Sarparanta u. a.: Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. In: Nat Genet. Band 44, Nr. 4, 26. Feb 2012, S. 450–455, S1-2. doi:10.1038/ng.1103.
Weblinks
- Zentrum für neuromuskuläre Erkrankungen an der Washington University in St. Louis: Limb-Girdle Muscular Dystrophy (Lgmd) Syndromes2 (ausführliche Informationen, histologische Bilder)