
| Klassifikation nach ICD-10 | |
|---|---|
| Q45.0 | Sonstige angeborene Fehlbildungen des Verdauungssystems |
| ICD-10 online (WHO-Version 2019) | |
Das Johanson-Blizzard Syndrom (JBS) ist eine seltene, manchmal tödlich verlaufende Erbkrankheit mehrerer Organsysteme, die durch eine gestörte Entwicklung von Bauchspeicheldrüse, Nase und Kopfschwarte gekennzeichnet ist und mit Intelligenzminderung, Hörverlust und Kleinwuchs einhergeht. Die Störung wird gelegentlich als ektodermale Dysplasie beschrieben und üblicherweise als eine erblich bedingte Erkrankung der Bauchspeicheldrüse betrachtet. Die Erkrankung ist nach den amerikanischen Kinderärzten Ann J. Johanson und Robert. M. Blizzard benannt, die diese Störung 1971 erstmals beschrieben haben.
Erbgang
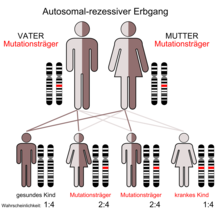
Das Johanson-Blizzard-Syndrom wird autosomal rezessiv vererbt. Das bedeutet, dass Mutationen auf beiden Allelen des Ubiquitin-Gens notwendig sind, um den entsprechenden Phänotyp zur Ausprägung zu bringen. Die Eltern der Betroffenen (Carrier) sind dabei üblicherweise phänotypisch gesund.
Pathophysiologie
Das JBS wird durch Mutationen im UBR1-Gen verursacht. UBR1 kodiert für eine Ubiquitin-Ligase. Ubiquitine sind in eukaryotischen Organismen universal exprimierte Proteine. Sie spielen eine Rolle in der posttranslationellen Modifikation anderer Proteine, indem sie sie bei Bedarf für den Abbau markieren. Ubiquitine funktionieren so, dass die Ubiquitin-Ligasen ein Ubiquitin kovalent an einen Lysinrest des (fehlerhaften) Zielproteins binden. Die Ubiquitin-Ligasen wiederholen diesen Prozess, sodass sich mit der Zeit eine Kette von Ubiquitinen an den Zielproteinen bildet. Man nennt dies Polyubiquination. Dies ist ein Signal für das Proteasom, das fehlerhafte Zielprotein abzubauen. Auf diese Weise spielt das Ubiquitin-Proteasom-System eine zentrale Rolle in der lysosomalen Degradation intracellulärer Proteine.
Aber Ubiquitine können auch an der posttranslationalen Modifikation normaler Proteine beteiligt sein. Beide Vorgänge, die posttranslationale Degradation und Modifikation sind in eukaryotischen Zellen wichtig für die Regulation zentraler Prozesse, wie der Zellteilung, bei Signalpfaden, der Apoptose, der Antwort auf Entzündungsreize und die Kontrolle von Entwicklungsprozessen.
Bei JBS führen die Mutationen des UBR1-Gens dazu, dass die Synthese der Ubiquitin-Ligase gestört oder unterbrochen ist. Bei gesunden Personen wird in den Pankreasdrüsengängen mehr UBR1 hergestellt als in allen anderen Zellen des Körpers. Die Funktionsstörung des Ubiquitin-Proteasom-Komplexes, die direkt auf die verminderte Produktion der Ubiquitin-Ligase zurückgeht, ist die unmittelbare Ursache für die chronisch-entzündlichen Prozesse im Pankreas, die mit dem Ersatz des Organstromas durch Fett- und Bindegewebe einhergeht und zu Fehlern in der Innervation der Acini und Langerhansschen Inseln führt. Sie ist auch verantwortlich für den Ausfall der Apoptose bei geschädigten Zellen und für eine fehlerhaften Proteinproduktion. Dies gilt ebenso für andere Gewebe, die anfällig sind für eine fehlerhafte UBR1-Expression, insbesondere das kraniofacial Gebiet, das Zentralnervensystem, die Skelettmuskelinnervation und die Zahnbildung.
Beim JBS hat man im UBR1-Gen Mutationen gefunden, die zum Kettenabbruch führen (Nonsense-Mutationen), solche die zu einem Austausch einer Aminosäure führen (Missense-Mutationen) sowie Splice-Site-Mutationen bei beiden Elternteilen von betroffenen Patienten. Dies bestätigt die Annahme einer homozygoten Natur des JBS-Phänotyps. Die variable Ausprägung des Phänotyps mit einer Rest-Ubiquitin-Ligase-Aktivität bei einigen Patienten wird mit dem Vorkommen sogenannter hypomorpher Mutationen bei einem Elternteil erklärt. Das UBR1-Gen liegt auf dem menschlichen Chromosom 15.
Klinisches Bild
Störungen der Pankreasfunktion
Das Kardinalsymptom des JBS ist die exokrine Pankreasinsuffizienz. Der damit einhergehende Mangel an Lipase, Trypsin, Trypsinogen und anderen Bestandteilen des Pankreassekretes führt insbesondere auch zu Fett-Malabsorption und stellt zusammen mit einer Störungen der endokrinen Pankreasfunktionen (gestörte Glucagon-Sekretion bei fehlenden Reaktion des Pankreas auf eine Hypoglykämie) die Hauptprobleme bei Patienten mit einem JBS dar. Bei JBS kann die exokrine Pankreasinsuffizienz, die zusammen mit entwicklungsbedingten Störungen der Apoptose im Pankreasgewebe, sowie pränataler und chronischer entzündlicher Schädigung der Pankreasdrüsengänge auftritt als Folge einer kongenitalen Verfettung der Pankreasdrüsengänge in Erscheinung treten. Es wurden Fälle berichtet, bei denen es zu einer vollständigen Verfettung des Pankreas gekommen ist. Dabei handelt es sich um einen fortschreitenden, manchmal tödlich verlaufenden Prozess.
Das JBS geht, wie bereits erwähnt, auch mit einer endokrinen Pankreasinsuffizienz einher. In den Langerhans-Inseln werden die Hormone Glucagon, Insulin und Somatostatin produziert. Beim JBS kann es zu einem bindegewebigen Umbau des Pankreas kommen, mit der Folge eines fettigen Ersatzes oder einer fehlerhaften Innervation der Inseln. Dies führt häufig zu einem Diabetes mellitus. In einem Fall wurde ursächlich dafür auch eine Insulinresistenz beschrieben. Die Produktion von Pankreassekret zusammen mit Elektrolyten und Bikarbonat ist bei Patienten mit JBS oft nicht beeinträchtigt.
Weitere endokrine Störungen
Beim JBS können auch noch andere endokrine Störungen auftreten. So wurden Hypothyreose,Mangel an Wachstumshormon und Hypophyseninsuffizienz beobachtet. Bei letzterem wurden Hamartome und Bildungsstörungen der Adenohypophyse als Ursache gefunden. Die Wachstumsstörung und der daraus folgende Minderwuchs beim JBS können eine Folge des Wachstumshormonmangels bei Hypophysenvorderlappeninsuffizienz und damit einhergehender Fettmalabsorption sein.
Nase
Die Fehlbildungen der Nasenflügel ist ein charakteristisches Merkmal des JBS. Es wird eine teilweise Anlage und auch vollständige Abwesenheit von Knorpel, Muskel- und Bindegewebe der Nase beschrieben. Dies resultiert in einem sehr ungewöhnlichen Aussehen der Nase bei den Patienten.
Nervensystem
Viele Patienten mit einem JBS zeigen eine Minderbegabung, die unterschiedlich stark ausgeprägt sein kann. Einzelne Fälle mit einer normalen Intelligenz und alterentsprechender sozialer Entwicklung wurden beschrieben.
Gehör
Veränderungen des Innenohres führen bei vielen Patienten mit einem JBS zu einer beidseitigen Innenohrschwerhörigkeit. Als Ursache wurden zystische Veränderungen von Cochlea und Gleichgewichtsorgan mit einer daraus folgenden Erweiterung beschrieben. Veränderungen des Schläfenbeins tragen zu den Hörschäden bei, weil dadurch Innervation und Entwicklung des Innenohrs beeinträchtigt werden.
Kraniofaziale Dysmorphien
Veränderungen, die Kopf, Gesicht, Kiefer (Anatomie) und Zähne betreffen werden unter dem Begriff kraniofaziale Dysmorphien zusammengefasst. Die beim JBS gefundenen Veränderungen sind vielgestaltig, sie betreffen folgende Organe, bzw. Körperteile: Defekte der Kopfschwarte mit unregelmäßigem Haarwuchs, dünne Kopfhaut, bis Aplasia cutis congenita, vergrößerte Fontanelle, Größenminderung des Kopfes (Mikrozephalie), gewölbte Stirn, Fehlen der Augenbrauen und Wimpern, Augenform ähnlich dem Downsyndrom, Fisteln des Tränennasenganges, abgeflachte Ohrmuscheln.,Mikrognathie (kleine Kiefer, wobei vorwiegend der Unterkiefer betroffen ist), angeborene Spaltbildung der Knochen in der Umgebung der Augenhöhle und fehlerhaft angelegte Milchzähne bei Fehlen der dauerhaften Zähne, oder einer Mikrodontie.
Störungen anderer Organsysteme
Weitere angeborene Störungen betreffen andere Organe. Die weniger häufig vorkommenden Störungen sind: Analatresie,Vesikorenaler Reflux; Doppelanlagen von Uterus und Vagina bei Mädchen,Neugeborenengelbsucht aufgrund einer Leberfibrose mit portaler Hypertension;Dilatative Kardiomyopathie,Dextrokardie,Atriumseptumdefekt und Ventrikelseptumdefekt;geringes Geburtsgewicht,Gedeihstörung,Muskelhypotonie; Angeborener Katarakt * und Café-au-lait-Flecke.
Diagnose und Differentialdiagnose
Übersichtsarbeiten verschiedener Autoren haben unabhängig von der Anzahl der bisher publizierten Fälle die exokrine Pankreasinsuffizienz als Kardinalsymptom beschrieben. Klinisch wird dies aufgrund des progressiven Charakters der Gewebeschädigung aber nicht immer von Anfang an beobachtet. Im Verlauf des ersten Lebensjahres der Kinder stellt sich üblicherweise ein typisches Bild eines Malabsorptionssyndroms mit voluminösen, fettigen Stühlen ein. Betrachtet man die am häufigsten vorkommenden Symptome, dann findet sich in etwa einem Dutzend Publikationen (n=38) die Kombination von Malabsorptionssyndrom aufgrund exokriner Pankreasinsuffizienz, Hypoplasie der Nasenflügel, Minderbegabung und Kopfhautläsionen. Ältere Arbeiten haben bei geringerer Fallzahl (n=22) zudem einen Minderwuchs als häufiges Symptom aufgelistet. In Fällen, in denen die Patienten keine oder nur diskrete weitere Zeichen haben, stellt sich dann die Differentialdiagnose einer angeborenen Pankreasinsuffizienz. Hier kommen außer dem JBS im Wesentlichen nur die Cystische Fibrose und das Shwachman-Bodian-Diamond-Syndrom (SBDS) in Frage. Falls aufgrund der sonstigen klinischen Befunde eine CF nicht infrage kommt, kann die Differentialdiagnose zum SBDS einfach anhand der beim JBS intakten Bicarbonat-Ausscheidung getroffen werden. Eine genetische Analyse bestätigt dann die Verdachtsdiagnose. In den weitaus überwiegenden bisher beschriebenen Fällen, kann eine Verdachtsdiagnose anhand der Kombination der Pankreasinsuffizienz zusammen mit den häufigsten Auffälligkeiten gestellt werden. Dies sind (in fast allen Fällen) die Hypoplasie oder Aplasie der Nasenflügel verbunden mit dem Vorliegen einer schmalen, langen Oberlippe, die den Patienten ein sehr charakteristisches Aussehen verleiht, sowie (seltener) Skalpdefekte, eine Innenohrschwerhörigkeit und eine Mikrozephalie. Minderwuchs und eine mentale Retardierung in sehr variablem Ausmaß können ebenfalls vorliegen.
Behandlung
Obwohl es keine Heilung für das JBS gibt, kann die Milderung einzelner Symptome durchaus erfolgreich sein. Dabei bestimmen der Schweregrad der Ausprägung einzelner Symptome die Notwendigkeit und die Erfolgsaussichten der ausgewählten Behandlungsverfahren. Die Pankreasinsuffizienz wird gewöhnlich mit Pankreatin substituiert, zudem stehen Rizoenzyme zur Verfügung. Die kraniofazialen und Skelett-Deformitäten können falls nötig chirurgisch korrigiert werden. Bei Hörminderungen können Hörhilfen verordnet und entsprechende Schulungen angezeigt sein. Patienten mit einer Minderbegabung profitieren von speziell für JBS-Erkrankte angepasste ergotherapeutische Maßnahmen.
Geschichte
Bereits im Jahre 1965 wurde das Krankheitsbild als Trypsinogenmangel durch den US-amerikanischen Arzt Philip L. Townes (18. Feb. 1927 – 1. Apr. 2017) beschrieben, dann weitere Betroffene durch die US-amerikanischen Kinderärzte M. D. Morris und D. A. Fisher, 1967. Die israelische Humangenetikerin Ruth Gershoni-Baruch und Mitarbeiter stellten fest, dass es sich bei den vorbeschriebenen Patienten um das Johanson-Blizzard-Syndrom handelte.
Siehe auch
Weblinks
- Johanson-Blizzard-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)