

Konzepte zur Überwindung der Blut-Hirn-Schranke ermöglichen es, dem Gehirn für therapeutische Zwecke Wirkstoffe zuzuführen. Die Blut-Hirn-Schranke ist eine dynamische Grenzfläche, die über Influx (Zufluss, wörtlich: Einströmen) und Efflux (Abfluss) kontrolliert, welche Nährstoffe, Arzneistoffe, Drogen, Xenobiotika und sonstige Verbindungen dem Gehirn zugeführt werden können. Dadurch gewährleistet sie dem Zentralnervensystem (ZNS) ein optimales Milieu.
Ihre Schutzfunktion macht die Blut-Hirn-Schranke jedoch auch zu einer Barriere für viele potenzielle Wirkstoffe und vereitelt so deren Einsatz in medikamentösen Therapien. Etwa 98 % der potenziellen Neuropharmaka scheitern daran. So lassen sich nur relativ wenige neurologische und psychiatrische Erkrankungen wie beispielsweise affektive Störungen wie Depressionen, Epilepsie oder chronische Schmerzen mit kleinen lipophilen Wirkstoffen behandeln.
Dagegen gibt es keine Therapie für neurodegenerative Erkrankungen wie die Alzheimer-Krankheit, Chorea Huntington und die Amyotrophe Lateralsklerose (ALS). Für Gehirntumoren, Schlaganfälle, Rückenmarksverletzungen und Schädel-Hirn-Traumata sind keine effektiven medikamentösen Therapien bekannt. Auch bei im Kindesalter auftretenden Syndromen wie Autismus, lysosomalen Speicherkrankheiten, dem Fragiles-X-Syndrom oder Ataxie stellt die Blut-Hirn-Schranke eine Barriere dar, die bisherige medikamentöse Therapieansätze verhindert. Selbst bei Erkrankungen wie Multipler Sklerose kann die Progression der Erkrankung im Zentralnervensystem nicht gestoppt werden, da die verabreichten Medikamente nur in der Peripherie wirken. Prinzipiell könnten viele dieser Erkrankungen mit Wirkstoffen, beispielsweise auf Basis von Enzymen, Genen oder biotechnologisch hergestellten Proteinen, behandelt werden – wenn sie die Blut-Hirn-Schranke überwinden könnten. Eine Therapie ist aber nur möglich, wenn diese Substanzen in ausreichender, das heißt therapeutisch wirksamer Konzentration auch an den Wirkort – also das Zentralnervensystem – gelangen können. Es wird daher seit Jahrzehnten intensiv an Methoden geforscht, die einen Wirkstofftransport in das Gehirn unter Umgehung oder – idealerweise selektiver – Öffnung der Blut-Hirn-Schranke ermöglichen sollen. Eine Reihe von Strategien zur Überwindung der Blut-Hirn-Schranke wurde dabei entwickelt oder befindet sich noch im Entwicklungsstadium.
Umgehen der Blut-Hirn-Schranke – intrathekale und intraventrikuläre Wirkstoffapplikation

Die naheliegendste Form des Wirkstofftransportes in das ZNS unter Umgehung der Blut-Hirn-Schranke stellt die Injektion direkt in den Liquor cerebrospinalis (intrathekal) oder direkt in die Hirnventrikel (intraventrikulär) dar. Der Wirkstoff wird dabei direkt in den Liquor injiziert. Angewendet wird dieses Verfahren beispielsweise als intrathekale Chemotherapie unter anderem mit dem Folsäure-Antagonisten Methotrexat (MTX), mit Cytarabin (AraC) und Cortisol; speziell bei Patienten mit akuter lymphatischer Leukämie und aggressiven Lymphomen. Die drei Wirkstoffe werden in der triple intrathecal chemotherapy zur Behandlung der Hirnhaut-Leukämie zusammen in den Liquor appliziert.
Die intrathekale Wirkstoffapplikation ist – verglichen mit der intravenösen (systemischen) Gabe von Wirkstoffen – deutlich aufwändiger und für viele Patienten auch unangenehmer. Darüber hinaus bestehen bei derartigen Darreichungsformen aufgrund der deutlich erhöhten Infektions- und Verletzungsgefahr besonders strenge Anforderungen an Hygiene und technische Fertigkeiten des Anwenders. Durch die Injektion von Wirkstoffen mit Depotwirkung (slow release) können die Behandlungsintervalle auf längere Zeiträume – beispielsweise 14-täglich – gestreckt werden. Weniger aufwändig ist die Verwendung eines Ommaya-Reservoirs, das unter die Kopfhaut implantiert wird. Einen ähnlichen Ansatz bieten implantierbare Medikamentenpumpen. Bei schweren Schmerzzuständen kann diese Methode beispielsweise für die Dosierung von Morphin gewählt werden. Auch zur Behandlung von Spastiken, beispielsweise bei Multipler Sklerose mit Baclofen, kann der Wirkstoff über eine solche Pumpe intrathekal appliziert werden. Die Methode wurde erstmals 1984 angewendet und ist seitdem etabliert.
Intrathekal applizierte Wirkstoffe werden meist speziell für diese Darreichungsform formuliert. Sie dürfen beispielsweise keine Bakterizide und eine Reihe anderer Hilfsstoffe enthalten, die in intravenös applizierten Medikamenten übliche Zusatzstoffe sind.
Für einige wenige Erkrankungen ermöglicht die intrathekale beziehungsweise die intraventrikuläre Wirkstoffapplikation eine wirksame Therapie. Für die Behandlung von Hirntumoren sind diese beiden Methoden zur Umgehung der Blut-Hirn-Schranke allerdings nicht geeignet. Die Ursache hierfür liegt in der auf nur wenige Millimeter begrenzten Diffusion der Wirkstoffe in das Parenchym des Gehirns.
Eine experimentell und therapeutisch nutzbare Lücke in der Blut-Hirn-Schranke sind die in das Gehirn eintretenden Hirnnerven. So konnte gezeigt werden, dass beispielsweise Neurotrophine, Neuropeptide, Insulin, Zytokine und sogar DNA, die über die Nase verabreicht wurden, über den Riechnerv in das Zentralnervensystem gelangen können. Ebenso konnte man über diesen Weg erfolgreich Stammzellen in das Gehirn einschleusen.
Überwindung der Blut-Hirn-Schranke für therapeutische Zwecke
Eine intakte Blut-Hirn-Schranke ist für jedes Wirbeltier lebensnotwendig. Für viele Wirkstoffe, die außerhalb des Zentralnervensystems ihre Wirkung entfalten sollen, ist die Retention an der Blut-Hirn-Schranke ein wichtiges Kriterium für die Zulassung, um die sonst zu erwartenden teilweise erheblichen Nebenwirkungen, insbesondere bei dauerhafter Einnahme eines Medikaments, sicher ausschließen zu können. Andererseits stellt die Blut-Hirn-Schranke bei der Behandlung neurologischer Erkrankungen für viele Verbindungen eine unüberwindliche Barriere dar.
Lipophilisierung

Die Strukturformel von Morphin

Codein (= 3-Methylmorphin) ist durch eine Methylierung etwas lipophiler als Morphin

Heroin ist durch zwei Acetylgruppen lipophiler als Morphin, weshalb es wesentlich leichter die Blut-Hirn-Schranke passieren kann.



Das Diffusionsvermögen eines Moleküls durch die Endothelien der Blut-Hirn-Schranke wird vor allem durch seine Fettlöslichkeit (Lipophilie) und Größe bestimmt. Durch eine Modifizierung des Moleküls mit lipophilen Gruppen kann deshalb eine verbesserte Gehirngängigkeit erreicht werden. Ein klassisches Beispiel hierfür ist die Di-Acetylierung des Naturstoffes Morphin zu Diacetylmorphin (Heroin). Heroin (log P=1,12) zeigt gegenüber Morphin (log P=0,2) eine über 25fach höhere Aufnahme im Gehirn (siehe dazu: Tabelle 1). Entsprechende Ergebnisse werden beim Brain-Uptake-Index (BUI) für radioaktiv markiertes Morphin, Codein und Heroin erhalten, das in die Halsschlagader injiziert wird. Für Morphin liegt der BUI unterhalb der Nachweisgrenze, bei Codein bei 24 % und für Heroin bei 68 %.
Dieses Prodrug-Konzept kann selbst bei peptidischen Wirkstoffen zu einer Verbesserung der Gehirngängigkeit führen.
Das Konzept versagt allerdings bei Molekülen mit einer molaren Masse größer als 500 g·mol−1, da solche Substanzen aufgrund ihrer Größe nicht mehr die Blut-Hirn-Schranke per Diffusion passieren können. Zudem geht mit der Lipophilisierung eine deutlich schlechtere Löslichkeit des Wirkstoffes einher. Bei der oralen Gabe können aber nur gelöste Wirkstoffe im Gastrointestinaltrakt aufgenommen werden. Die Lipophilisierung bewirkt natürlich auch eine erhöhte Aufnahme in anderen, nicht zerebralen, Zellen. Auch gegen Efflux-Transporter, die den eindiffundierten Wirkstoff wieder aus dem Endothel ausschleusen, ist die Lipophilisierung wirkungslos.
Ausnutzung der Transporter
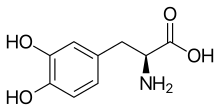

Im Endothel der Blut-Hirn-Schranke sind mehrere Transportsysteme, um das Gehirn mit essentiellen hydrophilen Substanzen zu versorgen. Ein Ansatz, Wirkstoffe in das Gehirn schleusen zu können, ist die Ausnutzung dieser Transporter. Dies wird beispielsweise bei der Therapie der Parkinson-Krankheit angewendet. Daran erkrankte Patienten haben im Gehirn einen Mangel des Neurotransmitters Dopamin. Die Gabe von Dopamin wäre diesbezüglich wirkungslos, da Dopamin die Blut-Hirn-Schranke nicht passieren kann. Verabreicht man dagegen Levodopa, eine nicht-proteinogene α-Aminosäure, so wird diese über den LAT1-Transporter dem Gehirn zugeführt und dort anschließend in Dopamin verstoffwechselt. Der LAT1-Transporter gehört zur Familie der LNAA-Transporter (large neutral amino acid).
Auch das Antiepileptikum Gabapentin, das Antihypertensivum α-Methyldopa und die Zytostatika Melphalan und Acivicin können über LNAA-Transporter die Blut-Hirn-Schranke passieren.
Die Obergrenze für die Ausnutzung der bestehenden Transportsysteme liegt bei einer molaren Masse von etwa 500 bis 600 g·mol−1.
Vektorisierung
Ein anderer Weg, um die Blut-Hirn-Schranke mit einem Wirkstoff zu überwinden, ist die Vektorisierung. Dieser Ansatz beruht auf der Beobachtung, dass einige Makromoleküle, wie Transferrin,Low Density Lipoprotein und Insulin über einen mehrstufigen, als rezeptorvermittelte Transzytose bezeichneten Prozess die Blut-Hirn-Schranke überwinden können. Über Rezeptoren, die sich an der Oberfläche der Endothelzellen der Hirnkapillaren befinden und in das Lumen der Blutgefäße hineinragen, werden die Makromoleküle in das Innere der Endothelzellen über Vesikel eingeschleust, um dann auf die andere Seite der Zelle (abluminale Seite) transportiert und ausgeschleust zu werden. Wird ein Wirkstoffmolekül an ein solches Makromolekül gebunden, kann die rezeptorvermittelte Transzytose zur Überwindung der Blut-Hirn-Schranke ausgenutzt werden.
Ein Beispiel hierfür ist der Transferrinrezeptor, der mit Hilfe gegen ihn gerichteter monoklonaler Antikörper zum Transport von Wirkstoffen durch die Blut-Hirn-Schranke genutzt werden kann. Dieser Rezeptor ist gewöhnlicherweise für den Transport von Eisen durch die Blut-Hirn-Schranke zuständig. Ein anderes Target ist der Insulinrezeptor, der auch von den Endothelzellen der Blut-Hirn-Schranke exprimiert wird. Mit beiden Vektoren wurden im Tiermodell verschiedene, auch größere, Peptide erfolgreich über die Blut-Hirn-Schranke geschleust. Speziell für die Therapie von neurodegenerativen Erkrankungen, für die nur geringe Wirkstoffkonzentrationen notwendig sind, ist die Vektorisierung ein vielversprechender Ansatz. Auch Zytostatika wie beispielsweise Doxorubicin wurden an Transferrinrezeptor-Antikörper gebunden.
Das Phänomen der Transzytose ist jedoch nicht auf Makromoleküle beschränkt. Wenngleich der genaue Mechanismus nicht immer geklärt ist, so konnte gezeigt werden, dass auch kleine Peptide und niedermolekulare Substanzen auf diese Weise in die Zelle gelangen und diese passieren können. Eine Vektorisierung zum Zweck der Passage der Blut-Hirn-Schranke ist somit auch mit kurzen Peptidsequenzen möglich. Als Vektoren für Wirkstoffe, wie beispielsweise Doxorubicin, fanden unter anderem basische Protegrin-Abkömmlinge, wie beispielsweise Syn-B, und das aus der Homöodomäne von Antennapedia, einem Transkriptionsfaktor von Drosophila, abgeleitete Penetratin Anwendung. Ein anderer Peptid-Vektor ist das aus elf überwiegend basischen Aminosäuren bestehende und aus der Transduktionsdomäne des HI-Virus isolierte HIV-TAT (engl. Trans-Activator of Transcription). Ein Peptid mit ähnlichen Eigenschaften ist das aus 27 Aminosäuren aufgebaute Transportan, ein zellpenetrierendes Peptid.
Mit transgenen Makrophagen können Proteine durch die Blut-Hirn-Schranke geschleust werden.
Kationisierung
Positiv geladene Moleküle (Kationen) können mit Hilfe der adsorptionsvermittelten Transzytose, auch kationischer Transport genannt, die Blut-Hirn-Schranke überwinden. Bei der adsorptionsvermittelten Transzytose bewirken elektrostatische Wechselwirkungen zwischen der durch Glykoproteine negativ geladenen Zelloberfläche und positiv geladenen Molekülen eine unspezifische Bindung an die Oberfläche von Zellen, in deren Folge eine Aufnahme und ein Transport durch das Zytoplasma der Endothelien erfolgt. Die kationische Transzytose durch das Endothel der Blut-Hirn-Schranke ermöglicht einen höheren Grad des Stofftransportes als die rezeptorvermittelte Transzytose.
Die Kationisierung von Antikörpern wurde in einer Reihe unterschiedlicher Studien und Anwendungsfeldern erfolgreich zur Passage der Blut-Hirn-Schranke eingesetzt. So beispielsweise, um β-Amyloidplaques sichtbar zu machen oder Mitochondrien zu targetieren.
Eine positive Ladung weisen bereits Peptide und Proteine auf, deren isoelektrischer Punkt im Basischen liegt. Ein Ansatz, die Aufnahme nicht basischer Peptide und Proteine im Gehirn zu verbessern, ist, diese mit Hilfe von natürlich vorkommenden Polyaminen, wie beispielsweise Putrescin, Spermidin oder Spermin, chemisch zu modifizieren. Eine Alternative dazu ist die im Kapitel Vektorisierung beschriebene Konjugation von Wirkstoffpeptiden und -proteinen an basische Peptide wie Syn-B. Auch synthetische Polyamine, wie beispielsweise Polyethylenimin, können zum erleichterten Transport von Wirkstoffen und DNA durch die Blut-Hirn-Schranke eingesetzt werden.
Der Effekt der Kationisierung ermöglicht zwar die Passage von Wirkstoffen und Diagnostika über die Blut-Hirn-Schranke, bewirkt aber gleichzeitig eine erheblich gesteigerte Aufnahme der applizierten Dosis in Leber und Nieren – mit den entsprechenden zu erwartenden Nebenwirkungen.
Nanopartikel



In den 1990er Jahren wurde in Versuchen mit Nanopartikeln, die aus biokompatiblen Polymeren aufgebaut sind, festgestellt, dass diese Partikel unter bestimmten Umständen in der Lage sind, die Blut-Hirn-Schranke zu passieren. Der Durchmesser dieser Partikel liegt üblicherweise bei 50 bis 300 nm. Die unfunktionalisierten, reinen Polymerpartikel sind in dieser Form nicht in der Lage durch das Endothel zum Gehirn transportiert zu werden. Der rezeptorvermittelte Transport ist nur durch eine spezielle Funktionalisierung, meist mit Polysorbat 80 oder Poloxameren, möglich. Als Polymere werden meist Polylactide (PLA), Polylactid-co-Glycolid (PLGA) und verschiedene Polycyanoacrylate, wie beispielsweise Polybutylcyanoacrylat (PBCA), verwendet, die pharmakologisch unbedenklich sind und für andere Anwendungen, beispielsweise als chirurgisches Nähmaterial, zugelassen sind. In die Partikel eingeschlossene Wirkstoffe können mittels rezeptorvermittelter Transzytose zum Gehirn transportiert werden.
Die wesentlichen Voraussetzungen für die Hirngängigkeit der Nanopartikel ist – neben ihrer Größe – eine möglichst lange Zirkulationszeit im Blut und die passende Oberflächencharakteristik. Die Plasmahalbwertszeit wird meist durch eine PEGylierung erreicht und die Wechselwirkung am Endothel mit dem bereits beschriebenen Polysorbat. Der genaue Transportmechanismus ist noch nicht endgültig geklärt. Der Polysorbat-Überzug der Partikel führt aber offensichtlich im Blutplasma zu einer Adsorption von Apolipoprotein E oder B an die Partikel. Dadurch werden die Nanopartikel als LDL-Mimetikum vom LDL-Rezeptor erkannt und in das Innere des Endothels transportiert. Danach wird der Wirkstoff entweder im Endothel freigesetzt, wodurch er per Diffusion zum Gehirn gelangen kann, oder die Partikel werden vollständig durch die abluminale Seite zum Gehirn ausgeschleust (Transzytose).
Der nanopartikuläre Wirkstofftransport ist derzeit noch in der präklinischen Forschung. Im Tiermodell (Ratte) wurden vielversprechende Ergebnisse bei der Behandlung von transplantierten Glioblastomen erzielt. Dabei wurden die Partikel mit Doxorubicin beladen. Der Transport von Doxorubicin in das Gehirn konnte dabei um den Faktor 60 gesteigert werden. Die wegen der weitgehenden Undurchlässigkeit der Blut-Hirn-Schranke für Chemotherapeutika nur schwer zu realisierende Chemotherapie bei Gehirntumoren ist eines der Hauptziele bei der Entwicklung dieser nanopartikulären Wirkstoff-Träger-Systeme.
Mit speziellen Liganden ist darüber hinaus die gewebe- beziehungsweise rezeptorspezifische Targetierung der Nanopartikel denkbar.
Neben dem nanopartikulären Ansatz mit Polymeren sind auch nanoskalige Liposomen und Dendrimere als potenzielle Wirkstofftransporter in der präklinischen Erprobung. Besondere Beachtung findet dabei auch die im Rahmen der gesamten Nanotechnologie stattfindende Diskussion über ihre Risiken.
Lösungsmittel und Tenside
Intravenös applizierte Verbindungen, wie Ethanol, Dimethylsulfoxid oder Glycerin, können zu einer lösungsmittelinduzierten Öffnung der Blut-Hirn-Schranke führen. Im Tiermodell (Küken) liegt dabei die Konzentration an Lösungsmittel oberhalb von 1 mg pro kg Körpergewicht. Diese Verbindungen stören vermutlich die Funktion der Zellmembran im Endothel, wodurch der Stofftransport durch transzelluläre Diffusion ermöglicht wird.
Werden kurzkettige Alkylglycerole, wie beispielsweise 1-O-Hexyldiglycerol, zusammen mit Marker-Substanzen in die Halsschlagader von Mäusen oder Ratten injiziert, so erhöht sich die Aufnahme dieser Marker im Gehirn signifikant. Größere Moleküle, die sonst nicht die Blut-Hirn-Schranke passieren, wie beispielsweise Methotrexat, Vancomycin oder Gentamicin, können – bedingt durch die Anwesenheit des Alkylglycerols – in das Gehirn diffundieren. Dieser Effekt wird bei der intravenösen Gabe von Alkylglycerol nicht beobachtet. Die amphipathischen Glycerole öffnen die Blut-Hirn-Schranke dabei für ungefähr 5 bis 120 Minuten. Die Konzentrationen der Alkylglycerole liegen im millimolaren Bereich. Offensichtlich bilden diese tensidähnlichen Verbindungen mit den Wirkstoffen, beziehungsweise Markern, vesikuläre Strukturen. Alkylglycerole sind weitgehend untoxisch und pharmakologisch unbedenklich. Der Mechanismus der Überwindung der Blut-Hirn-Schranke ist größtenteils noch ungeklärt. Es handelt sich aber offensichtlich um einen Transport durch die Tight Junctions.
Auch das Tensid Natriumlaurylsulfat erhöht bei der Injektion in die Halsschlagader die Durchlässigkeit der Blut-Hirn-Schranke deutlich. Natriumlaurylsulfat ist ein pharmakologischer Hilfsstoff, der in verschiedenen Wirkstoffformulierungen zur Anwendung kommt. Die entsprechende Applikation solcher Formulierungen kann daher zu unerwarteten Ergebnissen führen. So bewirkte der Hilfsstoff Natriumlaurylsulfat in einer Formulierung mit Interleukin-2, dass die Blut-Hirn-Schranke bei Katzen für die Markersubstanz Meerrettichperoxidase überraschend durchlässig wurde. Ähnliche Effekte wurden auch mit dem Hilfsstoff Polysorbat-80 beobachtet. Hierzu genügen bei einer Maus schon Dosen im Bereich von 3 mg pro kg Körpergewicht.Kyotorphin, ein neurophysiologisch aktives Dipeptid, ist nicht in der Lage die Blut-Hirn-Schranke zu passieren und eine neurologische Wirkung zu zeigen. Nur in Verbindung mit Polysorbat-80 wird die neurologische Wirkung erreicht.
Efflux-Inhibierung


Viele Moleküle sind sowohl wegen ihrer Größe als auch ihrer Lipophilie in der Lage die Blut-Hirn-Schranke zu passieren. Sie werden aber nach dem Diffundieren in das Zytoplasma der Endothelien durch Efflux-Pumpen, wie beispielsweise P-Glykoprotein, wieder zurück in das Lumen transportiert. Eine Strategie, um diese Moleküle dennoch dem Gehirn zugänglich zu machen, ist das Ausschalten dieser Efflux-Transporter. Prinzipiell ist dies möglich durch:
- Genregulation in der transkriptionalen oder translationalen Phase
- Veränderungen der Membran-Targetierung nach der Synthese der Transporter in den Ribosomen
- Unterbinden des Transportes durch Inhibitoren (Co-Drugs)
Während die ersten beiden Methoden sich noch in einem sehr frühen Entwicklungsstadium auf der Ebene von Zellkulturen befinden, liegen bei den Efflux-Inhibitoren ausgiebige Erfahrungen am Tier und aus klinischen Studien am Menschen vor.
Mittlerweile ist eine Reihe von Substanzen bekannt, die den Efflux – speziell durch P-Glykoprotein – inhibieren.
Mäuse, bei denen das MDR1-Gen abgeschaltet (Knockout) wurde, so dass im Endothel kein P-Glykoprotein produziert wird, zeigen für eine Reihe von Wirkstoffen eine signifikant erhöhte Aufnahme im Gehirn über die Blut-Hirn-Schranke. Im Vergleich zum Wildtyp der Maus stieg beispielsweise das Konzentrationsverhältnis Gehirn zu Blut bei den HIV-Protease-Inhibitoren Nelfinavir, Indinavir und Saquinavir um den Faktor 7 bis 36 an. Bei den Taxanen Docetaxel und Paclitaxel erhöht sich die Konzentration im Gehirn um den Faktor 7 bis 28 und bei Digoxin um den Faktor 10. Bei Verapamil wird die Aufnahme im Gehirn um den Faktor 8,5 verbessert.
Bei Wildtypen von Mäusen und Ratten, denen selektiv wirkende P-Glykoprotein-Inhibitoren, wie beispielsweise Valspodar (PSC 833, ein Ciclosporin-Derivat), Elacridar (GF120918) und Zosuquidar (LY335979), verabreicht wurden, konnten vergleichbare Ergebnisse erhalten werden. Bei Ratten, denen Ciclosporin verabreicht wurde, erhöht sich die Konzentration von Verapamil im Gehirn um den Faktor 9,6.
Verapamil – ein als Calciumantagonist zugelassenes Arzneimittel – ist im Tierversuch selbst ein wirksames Co-Drug, das die Aufnahme bei nachfolgend applizierten Wirkstoffen im Gehirn deutlich erhöhen kann. Dies wurde im Tiermodell unter anderem bei zytostatischen Vincaalkaloiden nachgewiesen. Eine ähnliche Wirkung zeigen Procyanidine.
Nachteilig bei dem Ansatz der Efflux-Inhibierung ist, dass die verabreichten Inhibitoren – speziell der ersten Generation, wie Verapamil und Ciclosporin – selbst pharmakologisch aktiv sind und so eine Reihe von unerwünschten Nebenwirkungen haben. Bei der zweiten und dritten Generation von P-Glykoprotein-Inhibitoren sind diese Effekte deutlich reduziert. Außerdem wird bei allen Zellen – die P-Glykoprotein exprimieren – selbiges inhibiert. So sind bei der systemischen Gabe von Efflux-Inhibitoren auch die apikale Seite der Darm-Epithelien, der Gallenkanälchen (Bilis canaliculi), der Nierenkanälchen und der Plazenta, sowie an der luminalen Seite die der Hodenkanälchen betroffen.
BCRP (Brustkrebs-Resistenz-Protein, Breast Cancer Resistance Proteine), der zweitwichtigste Efflux-Transporter der Blut-Hirn-Schranke, hat offensichtlich kaum einen Einfluss auf den Transport von Wirkstoffen. Dies wurde bei Versuchen an Knockout-Mäusen festgestellt, bei denen das BCRP-codierende ABCG2-Gen abgeschaltet wurde.
Die Efflux-Inhibierung wird insbesondere in der Krebstherapie verfolgt, da viele Krebszellen im Therapieverlauf P-Glykoprotein stark exprimieren und sich dadurch der Wirkung von Zytostatika weitgehend entziehen können. Die Tumoren sprechen dann nicht mehr auf die verabreichten Zytostatika an.
Öffnen der Blut-Hirn-Schranke für therapeutische Zwecke

Das Öffnen der Blut-Hirn-Schranke für therapeutische Zwecke ist, neben den beiden zuvor gezeigten Prinzipien, eine weitere Strategie, um Wirkstoffe dem Gehirn zuzuführen, die normalerweise nicht in der Lage sind die Blut-Hirn-Schranke zu passieren. Das Ziel dieser Verfahren ist eine möglichst reversible Öffnung oder zumindest Lockerung der Tight Junctions, um einen parazellulären Wirkstofftransport in das Gehirn zu ermöglichen. Mit dem zunehmenden Verständnis des molekularen Aufbaus der Blut-Hirn-Schranke – und hierbei vor allem der Tight Junctions – wurden neue Wege und Verfahren zur pharmakologischen, aber auch physikalischen, Öffnung der Blut-Hirn-Schranke entwickelt. Die meisten dieser Verfahren befinden sich noch in der präklinischen Erprobung.
Beim Öffnen der Blut-Hirn-Schranke besteht allgemein die Gefahr, dass für das Gehirn toxische Plasmaproteine eindiffundieren und dann chronische Neuropathologien auslösen können.
Tight-Junction-Modulation
Verbindungen, die einen Einfluss auf die Tight Junctions haben, werden als Tight-Junction-Modulatoren bezeichnet. Durch die Fortschritte im Bereich der genomischen Wirkstoffentwicklung, des High-Throughput Screening, der kombinatorischen Chemie und der Bioinformatik, wurde eine Reihe von Substanzen entwickelt beziehungsweise identifiziert, die in der Lage sind unmittelbar die einzelnen Peptide der Tight Junctions und Adherens Junction zu targetieren und damit den Zell-Zell-Kontakt der Endothelien zu modulieren.
Modulatoren, die unmittelbar die Tight Junctions targetieren, leiten sich beispielsweise von den Enterotoxinen der Bakterien Vibrio cholerae und Clostridium perfringens ab. Vibrio cholerae – ein Cholera-Erreger – bildet unter anderem das Zonula-Occludens-Toxin (ZOT, Zonula occludens = Tight Junction). ZOT ist ein aus 399 Aminosäuren aufgebautes, 45 kDa schweres Protein, das im Darm mit einem Oberflächenrezeptor – dem ZOT-Rezeptor – der dortigen Endothelien interagiert und dadurch eine intrazelluläre Signalkaskade auslöst, die noch nicht vollständig aufgeklärt ist. Es wird unter anderem das Enzym Proteinkinase A aktiviert, das den Abbau der Tight Junctions katalysiert. An Einzellagen zerebraler Endothelien bewirkt ZOT in vitro eine deutliche Reduzierung des transendothelialen elektrischen Widerstandes (TEER), die reversibel ist. Für die Markermoleküle Saccharose, Inulin, Paxlitaxel und Doxorubicin wird die parazelluläre Permeabilität signifikant erhöht. Auch das 12 kDa schwere aktive ZOT-Fragment ΔG sowie die aus nur sechs Aminosäuren (im Einbuchstabencode: FCIGRL) bestehende aktive ZOT-Domäne (AT1002) binden an den ZOT-Rezeptor.
Das aus 44 Aminosäuren bestehende OCC2-Peptid bindet selektiv an die zweite Domäne des Tight-Junction-Proteins Occludin, wodurch ebenfalls der parazelluläre Transport erleichtert wird.
Bradykinin, ein aus neun Aminosäuren aufgebautes gefäßerweiternd wirkendes Oligopeptid, bindet an die B2-Rezeptoren der luminalen Seite der Endothelien. Als Folge davon steigt die Konzentration an freien intrazellulären Calcium-Ionen und der mit den transmembranen Tight-Junction-Proteinen Occludin und Claudin verbundene Aktin-Myosin-Komplex wird aktiviert, wodurch die Tight Junctions geöffnet werden.
Osmotische Öffnung der Blut-Hirn-Schranke

Kurz nach der Entdeckung der Tight Junctions wurde 1970 die These aufgestellt, dass die Einwirkung von hyperosmotischen Lösungen auf die Endothelzellen die Blut-Hirn-Schranke öffnen könne. 1980 wurde diese Methode erstmals angewendet und 1984 wurde durch elektronenmikroskopische Aufnahmen der experimentelle Beweis für diese These erbracht. Elektronendichte Marker waren durch die Tight Junctions in das Gehirn diffundiert.
Über die Arteria carotis interna werden hyperosmolare Lösungen, beispielsweise von Mannitol oder Arabinose infundiert. Der unterschiedliche osmotische Druck zwischen den Endothelzellen und der infundierten Lösung bewirkt einen Flüssigkeitsverlust in den Endothelzellen, der zu deren Schrumpfung führt. Durch die Schrumpfung entstehen Zugkräfte zwischen den Zellen, was zu einer Öffnung der Tight Junctions und somit zur Öffnung der Blut-Hirn-Schranke führt.
Aufgrund des Konzentrationsgradienten zwischen intravasalem und interstitiellem Raum fließt in größerer Menge Wasser aus dem Plasma ins Gehirn zurück (bulk flow). Dadurch werden im Wasser gelöste Moleküle in das Gehirn eingeschwemmt, wobei ein Ödem entsteht.
Die durch die Schrumpfung der Endothelzellen bewirkte Öffnung der Tight Junctions beträgt etwa 20 nm. Dadurch können Moleküle mit einem hydrodynamischem Durchmesser von ebenfalls etwa 20 nm in das Gehirn eindiffundieren. Die Öffnung der Blut-Hirn-Schranke ist bei dieser Methode reversibel. Zehn Minuten bis spätestens zwei Stunden nach der Infundierung ist sie wieder vollständig hergestellt. Die Einwirkungszeit der hyperosmolaren Lösung beträgt etwa 30 Sekunden. Durch eine Vorbehandlung mit einem Na+/Ca2+-Kanalblocker kann die Öffnungsdauer der Blut-Hirn-Schranke verlängert werden.
Das Verfahren wurde im Tiermodell mit einer Vielzahl von wasserlöslichen Wirkstoffen, Peptiden, Antikörpern, Enzymen und viralen Vektoren für die Gentherapie getestet. Eine Reihe von klinischen Studien zur Therapie von Gehirntumoren in Kombination mit Chemotherapeutika werden in verschiedenen Kliniken durchgeführt. Die Ergebnisse sind für diese Anwendung vielversprechend.
Ultraschall





Die Blut-Hirn-Schranke lässt sich durch fokussierten Ultraschall öffnen. Dieser Effekt wurde erstmals 1956 nachgewiesen. Die Öffnung der Blut-Hirn-Schranke konnte durch die Anfärbung des Gehirns mit Trypanblau – einem Vitalfarbstoff, der normalerweise die Blut-Hirn-Schranke nicht passieren kann – und durch radioaktiv markiertes Phosphat nachgewiesen werden. Mikroskopisch konnten keine Veränderungen am Endothel beobachtet werden. Die Anwendung des Ultraschalls führte allerdings zu Hirnverletzungen. 1960 wurde dann erstmals die Blut-Hirn-Schranke mit nur einer geringen Schädigung des umliegenden Parenchyms durch Ultraschall geöffnet. Alle diese Versuche wurden mit hochintensivem fokussiertem Ultraschall, mit Leistungen im Bereich von 4000 Watt/cm², durchgeführt. Dabei entstehen Kavitationsblasen, die das Gewebe irreversibel zerstören können.
- Fokussierender Ultraschall mit Mikrobläschen
Die Öffnung der Blut-Hirn-Schranke mit Ultraschall und gleichzeitig applizierten Mikrobläschen (Microbubbles) kam 2001 zum ersten Mal zur Anwendung. Der Ansatz dabei ist, dass keine Kavitationsblasen generiert werden müssen, sondern injizierte Mikrobläschen die Funktion der sonst durch die hohe Ultraschallleistung erzeugten Kavitationsblasen übernehmen. Dadurch kann die Leistung des Ultraschalls deutlich reduziert werden; es besteht keine Gefahr mehr den behandelten Schädel, beziehungsweise das umliegende Gewebe, zu überhitzen. Die Technik ist mittlerweile so weit entwickelt, dass bei der Öffnung der Blut-Hirn-Schranke keine Apoptose, keine Ischämie oder sonstige Langzeitschädigung im Gehirn nachzuweisen sind. Wenige Stunden nach der Behandlung ist der alte Zustand der Blut-Hirn-Schranke wiederhergestellt.
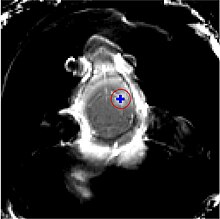
Der Fokus des Ultraschalls kann auf beliebige Areale im Gehirn gerichtet werden. Dadurch kann die Blut-Hirn-Schranke selektiv, auf bestimmte Hirnareale begrenzt, geöffnet werden. So können applizierte Wirkstoffe gezielt in diese Areale diffundieren. Die behandelten Areale lassen sich durch eine simultan laufende Magnetresonanztomographie (MRT) genau verfolgen. Dabei dringt das für die MRT verwendete Kontrastmittel, beispielsweise Gadopentetat-Dimeglumin, nur durch die geöffneten Areale der Blut-Hirn-Schranke in das Gehirn ein. Diese Bereiche werden dadurch im MRT deutlich sichtbar hervorgehoben. Das hochpolare Gadopentetat-Dimeglumin ist nicht in der Lage die ungeöffneten Bereiche der Blut-Hirn-Schranke zu passieren.
Im Tiermodell Maus werden bei der Anwendung von fokussiertem Ultraschall mit Mikrobläschen Frequenzen im Bereich von 0,5 und 2 MHz mit kurzen Pulslängen im Millisekundenbereich und Wiederholfrequenzen im Bereich von 1 Hz, über einen Zeitraum von weniger als einer Minute angewendet. Der optimale Frequenzbereich liegt unterhalb von 1 MHz. Die akustische Leistung beträgt weniger als ein Watt. Die verwendeten Mikrobläschen sind meist zugelassene Kontrastmittel aus der kontrastmittelverstärkten Sonographie. Sie haben typischerweise einen Durchmesser von 3 bis 4,5 µm, bestehen beispielsweise aus Humanalbumin und sind mit Perfluorpropan oder ähnlichen Schwergasen gefüllt.
- Mechanismus
Der Mechanismus zur Öffnung der Blut-Hirn-Schranke durch die Anwendung von fokussiertem Ultraschall, zusammen mit Mikrobläschen, ist noch nicht vollständig aufgeklärt. Die Wechselwirkung von Ultraschall und Mikrobläschen spielt dabei eine große Rolle und führt in vivo zu einer Reihe von biologischen Effekten. Eine wesentliche Rolle scheinen dabei Scherkräfte zu spielen, die durch Mikroströmungen erzeugt werden. Diese Mikroströmungen selbst kommen von Oszillationen der Mikrobläschen im Ultraschallfeld. Von den Endothelien selbst ist wiederum bekannt, dass sie auf Scherkräfte dynamisch reagieren können und Scherkräfte eine kritische Größe für die Homöostase sind. Elektronenmikroskopische Aufnahmen von Kapillargefäßen so behandelter Versuchstiere zeigen sowohl einen transzellulären als auch einen parazellulären Transport von entsprechenden Markermolekülen (Meerrettichperoxidase). Bei dem transzellulären Transport handelt es sich im Wesentlichen um Transzytose. Der parazelluläre Transport wird durch einen komplexen Desintegrationsprozess initiiert, bei dem die Tight Junctions ihre Funktion verlieren.
Die so geöffnete Blut-Hirn-Schranke ist durchlässig für niedermolekulare Chemotherapeutika, wie beispielsweise Doxorubicin und Antikörper, wie Trastuzumab. Auch die prinzipielle Machbarkeit des Transports von Genen in das Gehirn wurde mit dieser Methode im Tiermodell nachgewiesen. Das Verfahren zur Öffnung der Blut-Hirn-Schranke mit Ultraschall und gleichzeitig applizierten Mikrobläschen ist noch ein sehr junges Verfahren. Bisher wurde es nur an Versuchstieren erprobt. Bis zu einer möglichen Zulassung des Verfahrens am Menschen vergehen erfahrungsgemäß noch viele Jahre.
Die für die Bildgebung in der Diagnostik verwendete nicht-fokussierte Ultraschallstrahlung (Sonographie) beeinflusst die Integrität der Blut-Hirn-Schranke – auch bei der Gabe von Kontrastmitteln – nicht.
Literatur
- A. G. De Boer, W. Sutanto: Drug Transport Across the Blood-brain Barrier. CRC Press, 1997, ISBN 90-5702-032-7
- D. J. Begley u. a.: The Blood-brain Barrier and Drug Delivery to the CNS. Informa Health Care, 2000, ISBN 0-8247-0394-4
- P. Ramge: Untersuchungen zur Überwindung der Blut-Hirn-Schranke mit Hilfe von Nanopartikeln. Shaker Verlag, 1999, ISBN 3-8265-4974-0