
| Klassifikation nach ICD-10 | |
|---|---|
| E75.2 | Sonstige Sphingolipidosen Krabbe-Krankheit |
| ICD-10 online (WHO-Version 2019) | |
Der Morbus Krabbe (auch globoidzellige Leukodystrophie oder Globoidzell-Leukodystrophie) ist eine seltene angeborene monogenetische Stoffwechselstörung aus der Gruppe der Sphingolipidosen. Damit gehört die Erkrankung zu den lysosomalen Speicherkrankheiten.
Die Erkrankung wird autosomal rezessiv vererbt. Ursache ist ein Defekt im β-Galactocerebrosidase-Gen (GALC), welches für das Enzym Galactocerebrosidase kodiert. Dem Defekt können über 200 verschiedene Mutationen zugrunde liegen.
Der Mangel an dem Enzym Galactocerebrosidase führt zur Anreicherung des Cerebrosids Galactosylceramid in Makrophagen (Ausbildung von „Globoidzellen“). Die Anreicherung des Metabolits Psychosin führt weiterhin zur Schädigung der Oligodendrozyten. Die Fortsätze dieser Zellen bilden die Myelin-Markscheiden, welche die Axone der Nervenzellen umgeben. Durch die Schädigung der Oligodendrozyten kommt es zur Demyelinisierung der Nervenzell-Fortsätze, zu einer Entzündungsreaktion und dem bindegewebigen Umbau der weißen Substanz (Leukodystrophie).
Der Großteil der Fälle tritt im ersten Lebensjahr (infantile Verlaufsform) auf. Daneben gibt es eine Spätform (late-onset), die im späteren Kleinkindalter und anderen Lebensaltern auftritt. Es existiert keine kurative (heilende) Therapie. Die infantile Verlaufsform führt zum Tod im Kleinkindalter, die Prognose später auftretender Erkrankungen ist individuell unterschiedlich.
Die Erkrankung ist nach dem dänischen Neurologen Knud Krabbe benannt, der mehrere Fälle beschrieb.
Epidemiologie
Der Morbus Krabbe ist eine seltene Krankheit. Die Erkrankung tritt weltweit auf, wobei die Häufigkeit des Auftretens regional unterschiedlich ist. Auf 100.000 Lebendgeburten kommen durchschnittlich 1–2 von der infantilen Verlaufsform betroffene Kleinkinder (Inzidenz). Die anderen Verlaufsformen des Morbus Krabbe treten seltener auf, möglicherweise wird deren Inzidenz aber auch unterschätzt.
Genetik
Morbus Krabbe ist eine Erkrankung, die auf einem Gendefekt im β-Galactocerebrosidase-Gen (GALC) beruht. Das Gen liegt auf dem Chromosom 14 im Abschnitt q3.1. Das Genprodukt ist das Enzym Galactocerebrosidase, das Bestandteile der Myelinscheide von Nervenzellen abbaut.
Die durch Mutationen hervorgerufenen Defekte im GALC-Gen sind sehr heterogen. Bisher wurden über 200 verschiedene pathologische Genvarianten nachgewiesen, darunter große und kleine Deletionen, Frameshift-Mutationen und verschiedene weitere Punktmutationen. Die am häufigsten vorkommende pathologische Genvariante resultiert aus einer großen Deletion von 30 kb.
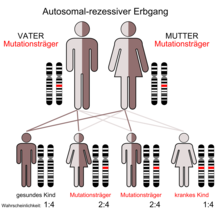
Die Erkrankung wird autosomal-rezessiv vererbt. Beide Eltern eines Betroffenen sind heterozygote Träger einer pathologischen GALC-Genvariante. Die Erkrankungswahrscheinlichkeit für ein Kind des Paares liegt bei 25 %.
Pathologie

Das Enzym Galactocerebrosidase spaltet in den Lysosomen den Einfachzucker Galactose von Galactocerebrosiden ab. Infolge von Defekten im β-Galactocerebrosidase-Gen ist beim Morbus Krabbe die Aktivität des Enzyms stark vermindert oder fehlend.
Der Mangel an dem Enzym Galactocerebrosidase führt zur Anreicherung des Cerebrosids Galactosylceramid in Makrophagen (Ausbildung von „Globoidzellen“). Weiterhin führt die Anreicherung von Psychosin zur Schädigung der Oligodendrozyten. Die Fortsätze dieser Zellen bilden die Myelin-Markscheiden, welche die Axone der Nervenzellen umgeben. Durch die Schädigung der Oligodendrozyten kommt es zur Demyelinisierung der Nervenzell-Fortsätze, zu einer Entzündungsreaktion und dem bindegewebigen Umbau der weißen Substanz (Leukodystrophie).
Symptome
Die Symptomatik beginnt meist im Alter von drei bis sechs Monaten. Die Kinder sind leicht irritierbar und neigen zu schwer beeinflussbaren Schreiattacken. Die kognitiv-motorische Entwicklung kommt zum Stillstand. Auf äußere Reize hin kann es zu tonischer Streckung der Beine kommen. Reflexe sind nicht mehr auslösbar. Bei Optikusatrophie kommt es zu Blindheit.
Im weiteren Verlauf entwickeln sich Taubheit, ein permanenter Opisthotonus mit gebeugten Armen und gestreckten Beinen sowie Hypersalivation und Fieber. Bei den spät beginnenden Sonderformen kommt es zu denselben Symptomen, nur beginnt die Krankheit später und verläuft auch langsamer.
Diagnose
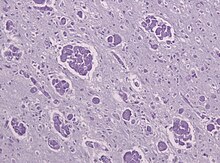
Im Hirnwasser (Liquor cerebrospinalis) findet sich typischerweise eine erhöhte Eiweißkonzentration. Die Nervenleitgeschwindigkeit ist vermindert. In bildgebenden Verfahren zeigt sich die Demyelinisation. In Autopsiepräparaten zeigen sich Monozyten und mehrkernige Makrophagenansammlungen (globoid cells) mit PAS-positiven, aber nicht metachromatischen Einschlüssen in gliotisch verändertem Hirngewebe.
Um die Diagnose zu sichern, kann man die Aktivität des Enzyms Galaktocerebrosidase in Leukozyten oder Fibroblastenkulturen bestimmen oder eine Genanalyse durchführen. Bei letzterer findet sich eine Mutation im Chromosom 14 (Abschnitt q3.1).
Da es sich um eine rezessiv vererbte Krankheit handelt, beträgt das Risiko für Geschwister ebenfalls zu erkranken 25 %. Durch pränatale Diagnostik kann man einen Morbus Krabbe schon in utero (in der Gebärmutter) diagnostizieren bzw. ausschließen. Ebenso kann man Verwandte untersuchen, ob sie Überträger der Erkrankung sind.
Therapie
Die Erkrankung ist nicht heilbar. Entsprechend beschränken sich die Behandlungsmöglichkeiten von an der infantilen Verlaufsform erkrankten Kindern, die bereits Zeichen der Erkrankung zeigen, auf die symptomatische Therapie bzw. palliativmedizinische Versorgung. Die nachfolgend beschriebenen Maßnahmen können auch bei der Late-onset-Verlaufsform notwendig sein.
Mit physiotherapeutischen Maßnahmen kann die Spastik der Muskulatur günstig beeinflusst werden. Eine Verbesserung der Spastik kann weiterhin durch die Gabe von Clonazepam oder des Muskelrelaxans Baclofen erreicht werden. Auch die therapeutische Anwendung von Botulinumtoxin an ausgewählten Muskelgruppen wurde beschrieben.
Es muss auf ausreichende Energie- und Flüssigkeitszufuhr geachtet werden. Bei und nach dem Füttern sollte eine aufrechte Körperposition beibehalten werden, um Reflux und Erbrechen vorzubeugen. Im weiteren Verlauf müssen Flüssigkeiten häufig angedickt werden, weiterhin kann die Anlage eines Gastrostomas notwendig werden. Bei Verstopfung (Obstipation) sind Abführmittel (Laxanzien) indiziert. Die Darmmotilität kann durch Erythromycin unterstützt werden.
Zur Behandlung von neuropathischen Schmerzen und dem Schutz vor Krampfanfällen wird Gabapentin angewendet. Zum Durchbrechen von Krampfanfällen ist die rektale Gabe von Diazepam sinnvoll.
Bei den Betroffen sollte die Ansammlung von Restharn vermieden werden, um Harnwegsinfekte zu vermeiden. Zur vollständigen Harnblasenentleerung kann eine vorübergehende Katheterisierung notwendig sein. Die dauerhafte Anlage von Kathetern ist aufgrund des Infektionsrisikos nicht empfohlen.
Wird die infantile Verlaufsform der Erkrankung vor dem Auftreten von Symptomen diagnostiziert, so kann eine Stammzelltransplantation versucht werden. Eine frühzeitige Therapie innerhalb der ersten Lebenswochen kann die Lebenserwartung verlängern und die funktionelle Entwicklung verbessern.
Prognose
Kinder mit der infantilen Verlaufsform des Morbus Krabbe versterben nach Symptombeginn meist in den ersten zwei Lebensjahren. Eine Stammzelltransplantation vor Auftreten von Erkrankungszeichen kann sowohl die Lebenserwartung verlängern als auch die funktionelle Entwicklung verbessern.
Die Prognose der Late-onset-Verlaufsform ist sehr variabel.
Früherkennung
Mehrere US-Bundesstaaten führen ein Neugeborenenscreening auf die Erkrankung durch. Ziel ist dabei, betroffene Neugeborene schnellstmöglich zu identifizieren und einer Stammzelltransplantation innerhalb des ersten Lebensmonats zuzuführen. Aus den gewonnenen Erfahrungen wurden mittlerweile Leitlinien-Empfehlungen zusammengefasst.
Forschungsgeschichte
Der dänische Neurologe Knud Krabbe beschrieb 1916 in der Zeitschrift Brain fünf Fälle einer fortschreitenden, tödlich verlaufenden Erkrankung bei Säuglingen, die durch eine Erhöhung des Muskeltonus, Entwicklungsrückstände, Fieberschübe und Opisthotonus gekennzeichnet ist. Dabei handelte es sich um die infantile Form der Globoidzellleukodystrophie, die ab den 1940er Jahren nach ihrem Erstbeschreiber benannt wurde.
Die verminderte Aktivität des Enzyms β-Galaktozerebrosidase bei der Erkrankung wurde Anfang der 1970er Jahre entdeckt. Ab 1980 war ein Mausmodell für die Erkrankung verfügbar (twitcher mouse). Die die β-Galaktozerebrosidase kodierende cDNA wurde 1993/1994 kloniert, in einer der Veröffentlichungen wurde zudem eine zum Morbus Krabbe führende Nonsense-Mutation beschrieben. Die Struktur des Gens wurde 1995 dargestellt. In diesem Jahr wurde auch die 30kb-Deletion, die bei vielen Patienten vorliegt, identifiziert.
Literatur
- Orsini et al.: Krabbe Disease. In: GeneReviews, 2018.
- David A. Wenger, Mohammad A. Rafi, Paola Luzi: Krabbe disease: One Hundred years from the bedside to the bench to the bedside. In: Journal of Neuroscience Research. Band 94, Nr. 11, 2016, S. 982–989, doi:10.1002/jnr.23743, PMID 27638583.
- Alfried Kohlschütter: Lysosomal leukodystrophies: Krabbe disease and metachromatic leukodystrophy. In: Handbook of Clinical Neurology. Band 113, 2013, S. 1611–1618, doi:10.1016/b978-0-444-59565-2.00029-0, PMID 23622382.
- Maria L. Escolar et al.: Clinical management of Krabbe disease. In: Journal of Neuroscience Research. Band 94, Nr. 11, 2016, S. 1118–1125, doi:10.1002/jnr.23891, PMID 27638597.
Weblinks
- Morbus Krabbe. In: Orphanet (Datenbank für seltene Krankheiten).
- Morbus Krabbe. In: Online Mendelian Inheritance in Man. (englisch)
- Morbus Krabbe bei der Europäischen Vereinigung gegen Leukodystrophien (ELA)