

Die Neuroonkologie (von altgriechisch νεῦρον neuron, deutsch ‚Nerv‘, ὄγκος onkos, deutsch ‚Anschwellung‘ und -λογία -logia, deutsch ‚Lehre‘, ‚Wissenschaft‘) ist eine Schwerpunktbildung der Medizin, die Neurologie und Onkologie verbindet. Es gibt im deutschsprachigen Raum keinen Facharzt und keine Zusatzbezeichnung für Neuroonkologie.
Die Neuroonkologie befasst sich vorwiegend mit der Diagnose, Therapie und Forschung von Tumorerkrankungen des Nervensystems, wozu die folgenden Tumorarten gezählt werden:
- primäre Tumoren des Zentralnervensystems (Hirntumore und Tumoren des Rückenmarks)
- sekundäre Tumoren des Zentralnervensystems (Hirnmetastasen und Metastasen des Rückenmarks)
- maligne Lymphome des Zentralnervensystems
- Tumoren des peripheren Nervensystems
Diese Aufzählung zeigt bereits, dass dies ein breites und schwierig abzugrenzendes Gebiet ist. Während die Behandlung auch zentralnervöser Lymphome den hämatologischen Behandlungsrichtlinien folgt, und Hirnmetastasen nicht abgekoppelt von der Grundtherapie des Primärtumores behandelt werden, sind periphere Nerventumoren gutartig, wie Neurinome, und wie andere Weichteiltumoren oftmals im orthopädischen Bereich angesiedelt.
Allgemeine Informationen
Primärtumoren des Zentralnervensystems
Primäre Hirntumoren können in jedem Alter auftreten, vom Säuglingsalter bis spät im Leben. Faktoren wie Alter, Tumorlokalisation und klinisches Erscheinungsbild sind bei der Differentialdiagnose hilfreich. Die meisten Arten von primären Hirntumoren treten häufiger bei Männern auf, mit Ausnahme von Meningeomen, die bei Frauen häufiger auftreten.
Metastasierende Tumoren des Zentralnervensystems
Die direkte Invasion oder Kompression von kontinuierlichen Geweben hängt mit der Nähe des Nervensystems zu anderen Strukturen zusammen.
Intrakranielle Metastasierung
Es gibt drei Arten von intrakraniellen Metastasen: Hirnmetastasen, Dural-Metastasen und Meninges-Leptomeningeal-Metastasen. Die Hirnmetastasierung kann einfach oder mehrfach sein und einen beliebigen Teil des Gehirns betreffen. Die Metastasierung von Durastrukturen erfolgt im Allgemeinen durch hämatogene Ausbreitung oder direkte Invasion von einem angrenzenden Knochen. Duralmetastasen können in das zugrunde liegende Gehirn eindringen und fokale Ödeme und damit verbundene neurologische Symptome verursachen. Diese Prozesse neigen aufgrund ihrer kortikalen Lage dazu, zu Beginn des Verlaufs Anfälle zu verursachen. Die Metastasierung der Leptomeninges ist eine seltene, aber allgemein anerkannte klinische Erscheinung bei Krebspatienten. Leptomeningeale Metastasen sind am häufigsten auf Brust-, Lungen- oder Melanom-Primärtumoren zurückzuführen.
Schädelmetastasen
Metastasen zum Schädel werden in zwei Kategorien unterteilt: Calvarium und Schädelbasis.
Primärtumoren des Zentralnervensystems
Eine erste Unterteilung von Tumoren des Zentralnervensystems (ZNS) findet zwischen primären (direkt aus dem ZNS stammend) und metastatischen (aus einem anderen Organ stammend) Tumoren statt. Letztere haben eine Inzidenz, die in etwa dem Zehnfachen der ersteren entsprechen. Hirntumoren sind Neoplasien, die sich im Hirn entwickeln. Tumoren wie das Meningeom, die aufgrund ihrer Raumforderung das Hirn komprimieren, aber nicht eindringen, sowie Hypophysen- und Epiphysentumoren, die am Hirnstamm liegen, werden fälschlicherweise immer wieder als Hirntumoren bezeichnet. Der Begriff intrakranielle Tumoren fasst diese exakter zusammen.
Primäre ZNS-Tumoren umfassen eine Vielzahl pathologischer Einheiten, von denen jede ihre eigene Naturgeschichte hat. Aufgrund der Tatsache, dass gliale Tumoren allein fast 40 Prozent dieser Tumoren ausmachen, kann zunächst zwischen glialen Tumoren (Gliomen) und nichtglialen Tumoren unterschieden werden. Die häufigsten Gliome sind Astrozytome (die aus den Astrozytenzellen der Glia stammen), Oligodendrogliome (aus Oligodendrogliazellen) und Ependymome (aus Ependymzellen).
Epidemiologie
Maligne Primärtumoren des Zentralnervensystems sind relativ selten und machen etwa 2 Prozent aller bösartigen Neoplasien aus. Tumorerkrankungen des Zentralen Nervensystems verteilen sich zu 95 Prozent auf das Gehirn und zu 5 Prozent auf Hirnhäute, Hirnnerven und Rückenmark. Sie können in jedem Lebensalter auftreten, das Erkrankungsrisiko steigt mit zunehmendem Alter. Bei Erwachsenen finden sich histologisch vom Stützgewebe der Nervenzellen ausgehende Gliome, wovon etwa 75 Prozent Glioblastome bzw. Astrozytome IV. Grades mit ungünstiger Prognose sind. Bei Säuglingen und Kleinkindern überwiegen embryonale Tumoren. In Deutschland erkrankten 2016 etwa 3.970 Männer und 3.460 Frauen an bösartigen Tumoren des Zentralen Nervensystems. Im Durchschnitt liegen die Überlebensraten bei 21 Prozent für Männer und 24 Prozent für Frauen. In Statistiken werden selten auch histologisch gutartige ZNS-Tumoren betrachtet, die bei etwa 6.000 Neuerkrankungen pro Jahr liegen. Etwa 65 Prozent davon gehen von den Hirnhäuten aus. Deutlich häufiger hiervon sind Frauen betroffen. Wenn sie nicht chirurgisch oder durch Strahlentherapie behandelt werden, können auch gutartige Tumoren aufgrund des fortschreitenden Wachstums im geschlossenen Schädelraum tödlich sein. Der häufigste bösartige intrakranielle ZNS-Tumor ist das Glioblastom, der häufigste gutartige das Meningeom.
Ätiologie
Für Neoplasien des Zentralen Nervensystems ist eine genetische Veranlagung relativ selten, obwohl einige Gliome als Komplikationen mehrerer familiärer Erkrankungen auftreten können.
Die Mutation einiger Tumorsuppressorgene charakterisiert mehrere erbliche Syndrome, die eine erhöhte Anfälligkeit für die Entwicklung von Hirntumoren zeigt. Folgende Mutationen und ihre Syndrome sind mit einem höheren Risiko für eine Entwicklung von Hirntumoren verbunden: NF1-Genmutation mit Typ-1-Neurofibromatose, APC-Mutation mit Turcot-Syndrom, PTCH-Mutation mit Gorlin-Syndrom und TP53- oder CHEK2-Mutation mit Li-Fraumeni-Syndrom.
Schwer zu identifizieren sind Umweltfaktoren im Zusammenhang mit primären Hirntumoren. In einigen Studien wurde die Exposition gegenüber Vinylchlorid mit einer erhöhten Inzidenz von hochgradigen Gliomen in Verbindung gebracht. Die einzige seltene, aber gut identifizierte Ursache für einen primären Hirntumor ist ionisierende Strahlung. Insbesondere die Strahlentherapie von Kindern mit Tinea capitis und von Patienten mit akuter lymphatischer Leukämie, Kraniopharyngeom oder Non-Hodgkin-Lymphom ist mit einem erhöhten Gliomrisiko verbunden. Bei AIDS-Patienten besteht ein erhöhtes Risiko für ein primäres Hirnlymphom.
Klinik
Anzeichen und Symptome
Die Symptome der zerebralen Neoplasie werden durch Verdrängung bzw. Zerstörung umliegenden Gewebes sowie Infiltrierung desselben verursacht.
Das häufigste Symptom, über das 35 Prozent der Patienten klagen, ist Kopfschmerz. Häufig charakteristisch ist das Auftreten schwerer Kopfschmerzen bei Patienten, die sonst selten darunter leiden, insbesondere wenn die Kopfschmerzattacken oder Migräne am Morgen stärker sind und von Übelkeit, Erbrechen und neurologischen Ausfällen begleitet werden. Bei Patienten, die häufiger unter Kopfschmerzen leiden, kann eine Veränderung der Form, die Zunahme der Häufigkeit oder Intensität der Anfälle ein Symptom für die Entwicklung eines Hirntumors sein. Bei etwa einem Drittel der Gliompatienten treten Krampfanfälle auf, insbesondere bei niedriggradigen oder ZNS-Tumoren. Fokale neurologische Ausfälle hängen mit dem Ort des Tumors zusammen. Bei 15 bis 20 Prozent der Gliompatienten kommt es auch zu Veränderungen des psychischen Status.
Bildgebende Diagnostik


Durch Computertomographie (CT) und Kernspinresonanztomographie (MRT) kann eine Neoplasie im Gehirn effektiv nachgewiesen werden. Zur Identifizierung von Läsionen ist die MRT empfindlicher als die CT, weist aber für Patienten mit Herzschrittmachern, inkompatiblen Prothesen, Metallklammern und anderem Kontraindikationen auf. Die CT bleibt die Methode der Wahl, um Verkalkungen innerhalb der Läsionen oder Knochenerosionen der Schädeldecke oder -basis zu erkennen. Die Verwendung von Kontrastmitteln, die im Fall der CT jodiert und im Fall der MRT paramagnetisch (Gadolinium) sind, ermöglicht die Erfassung von Informationen über die Vaskularisation und Integrität der Blut-Hirn-Schranke, eine bessere Definition der Tumorgeschwulst im Vergleich zum umgebenden Ödem und die Erstellung von Hypothesen über den Grad der Malignität. Die radiologische Untersuchung ermöglicht auch eine Bewertung der mechanischen Auswirkungen und die daraus resultierenden Veränderungen der Gehirnstrukturen, die sich durch den Tumor ergeben, wie zum Beispiel Hydrocephalus und Hernien, deren Auswirkungen tödlich sein können. Schließlich kann mit dieser Diagnostik in Vorbereitung einer Operation der Ort der Läsion oder die Infiltration des Tumors in lebenswichtige Bereiche des Gehirns bestimmt werden. Zu diesem Zweck ist die MRT effizienter als die CT, da sie dreidimensionale Bilder liefern kann.

Diagnostische radiologische Bildgebungsinstrumente heben die Veränderung des neoplastischen Gewebes im Vergleich zum normalen Gehirnparenchym hervor (durch Änderungen der elektronisch dargestellten Dichte des Gewebes bei der CT und der Signalintensität bei der MRT). Wie die meisten pathologischen Gewebe sind auch Tumore durch eine erhöhte Ansammlung intrazellulären Wassers erkennbar. Im Computertomogramm erscheinen sie hypodens, das heißt von geringerer Dichte als das Gehirnparenchym, im Kernspinresonanztomogramm bei Spin-Gitter-Relaxation hypointens und bei Spin-Spin-Relaxation sowie Protonengewichtung (PD) hyperintens.

Auf einer radiologischen Aufnahme sollte der gesunde Gehirnbereich keine besondere Lumineszenz aufweisen. Daher ist es selbstverständlich, dass auf größere Kontrastsignalbereiche geachtet wird.
Im Tumorgewebe ist im Allgemeinen der größere Anteil der Kontrastverstärkung auf die besondere Blut-Tumor-Schranke zurückzuführen, die den Durchgang von Iod (CT) und Gadolinium (MRT) in den intratumoralen extravaskulären Interstitialraum ermöglicht. Dadurch steigt das Signal (Dichte oder Intensität) des Tumors. Es sollte jedoch darauf geachtet werden, dass die Kontrastverstärkung die Neoplasie von Periwundödemen nicht mit Sicherheit abgrenzt. Tatsächlich zeigt der anatomisch-pathologische Befund bei malignen infiltrierenden Gliomen Tumorgewebe, wie zum Beispiel beim Glioblastom und anaplastischem Astrozytom, auch jenseits des vasogenen Ödems, das durch die Zerstörung der Blut-Hirn-Schranke durch den Tumor verursacht wird. Letzterer klinischer Zustand ist durch diagnostische Bildgebung schlecht nachweisbar.
Die Computertomographie des Gehirns zeigt typischerweise eine Gewebsmasse, die entweder durch Kontrast verstärkt werden kann. Bei der CT erscheinen niedriggradige Gliome normalerweise isodens zum normalen Parenchym und zeigen daher möglicherweise keine Kontrastverstärkung. In ähnlicher Weise sind Läsionen in der Fossa cranii posterior, der hinteren Schädelgrube, im CT schwer zu identifizieren. Folglich sind die Ergebnisse einer solchen Tomographie allein nicht immer für diagnostische Zwecke ausreichend. In zweifelhaften Fällen ist die Verwendung der empfindlicheren Kernspintomographie unerlässlich.
Auf -Kernspintomogrammen erscheint ein intrakranieller Tumor als massive Läsion, die nach Verwendung des Kontrastmittels lumineszierender werden kann. Eine Signalanomalie gibt es jedoch immer in -Kernspintomogrammen, die auf das Vorhandensein einer Neoplasie oder eines vasogenen Ödems hinweist. Normalerweise ist eine stärkere Lumineszenz (Kontrastverstärkung) ein Hinweis auf einen Tumor höheren Malignitätsgrades. Ein Kontrastring ist charakteristisch für ein Glioblastom, wobei der Lumineszenzanteil dem lebenswichtigen Teil des bösartigen Tumors und der dunklere -hypointense Bereich der Gewebenekrose entspricht.
Stadienbestimmung
Die meisten primären intrakraniellen Tumoren bleiben im Schädel lokalisiert, so dass systemische Staging-Verfahren nicht erforderlich sind.
Primäre neuroektodermale Tumoren, Medulloblastome, ZNS-Keimzelltumoren und primäre ZNS-Lymphome breiten sich dagegen häufig über den Subarachnoidalraum bis zu den Leptomeningen aus. Für alle Patienten mit solchen Diagnosen ist daher auch eine spinale Kernspintomographie bzw. eine Lumbalpunktion erforderlich.
Tumorarten
Gliome
Primäre Tumoren des Zentralnervensystems (ZNS) umfassen eine Vielzahl pathologischen Gewebes, von denen jedes seine eigene Naturgeschichte hat. Aufgrund der Tatsache, dass Gliome allein fast 40 Prozent aller ZNS-Tumoren ausmachen, ist es in der Literatur üblich, zwischen glialen und nichtglialen Tumoren zu unterscheiden.
Astrozytome
Zur Abstufung der Malignität von Astrozytomen wurden in der Literatur im Laufe der Zeit verschiedene Kategoriesysteme vorgeschlagen. Seit 1993 ist das von der Weltgesundheitsorganisation (WHO) vorgeschlagene vierstufige Bewertungssystem das am weitesten verbreitete und angewandte. Es basiert auf vier histologische Merkmale: erhöhte Zelldichte, Mitose, Endothelproliferation und Nekrose. Danach sind Astrozytome I. Grades, wie pilozytische Astrozytome, typischerweise gutartiger Histologie. Astrozytome II. Grades (diffus) weisen als einziges histologisches Merkmal eine erhöhte Zelldichte auf und sind Neoplasien geringeren Infiltrationsgrades. Eine signifikante Mitose zeigen Astrozytome III. Grades (anaplastisch). Und eine Endothelproliferation oder Nekrosen sind bei Astrozytomen IV. Grades, den sogenannten Glioblastomen, erkennbar.
Niedriggradige Astrozytome

Pilozytische Astrozytome (unter anderem das pilomyxoide Aastrozytom), subependymale Riesenzellastrozytome und pleomorphe Xanthastrozytome gehören zu den umschriebenen Tumoren. Diese sind etwas seltenere Neoplasien gutartiger Histologie, die oftmals nur durch eine Operation geheilt werden können. Sollte die Exzision unvollständig erfolgen, könnte das verbleibende tumoröse Gewebe erfolgreich mit einer Strahlentherapie behandelt werden. In seltenen Fällen, bei denen die die lokale Behandlung nicht anschlägt, kann eine systemische Chemotherapie erfolgreich sein, die individuell eingestellt sein muss. Kinder sprechen auf eine Kombination von Carboplatin und Vincristin an.


In der Computertomographie erscheinen diffuse Astrozytome II. Grades als schwächer intensive Läsionen. Bei der bevorzugten Kernspintomographie können Kontrastmittel diese Neoplasien möglicherweise nicht hervorheben, ihre Lumineszenz kann dünner und schwächer ausfallen. Eine intensivere kann auf Gewebe erhöhter Anaplasie hinweisen. Wann immer möglich, wird eine Biopsie vorgeschlagen, um Proben aus dem anaplastischen Teil des Tumors zu erhalten.
In den meisten Fällen sind Patienten mit diffusen Astrozytomen 20 bis 40 Jahre alt. Typisch ist bei ihnen das Auftreten epileptischer Anfälle. Bedingungen für eine günstige Prognose sind junges Alter, Tumorgröße unter 50 Millimeter und eine möglichst umfangreiche chirurgische Resektion des Tumors. Späte Rückfälle sind relativ häufig, weshalb die Patienten nach dem Entfernen des Tumors 15 Jahre lang nachuntersucht werden müssen.
Trotz ihres relativ trägen Verlaufs entwickeln sich die meisten Astrozytome zu Läsionen, die durch eine größere Anaplasie gekennzeichnet und normalerweise mit Operationen und Strahlentherapien nicht heilbar sind. Die Therapie für Patienten mit diffusen niedriggradigen Astrozytomen zeigt jedoch in der Literatur keinen einstimmigen Konsens. Die Rolle der vollständigen Resektion wird in Fachkontexten diskutiert. Die Ergebnisse einiger Studien zeigen, dass eine maximale Tumorentfernung die besten Ergebnisse liefert. Tatsächlich können kleine und einseitige Tumoren vollständig entfernt werden, wenn keine kritischen Strukturen des Gehirns beteiligt sind. Ein pragmatischer, für die Allgemeinheit der Fälle insgesamt akzeptabler Ansatz ist eine möglichst weitreichende Entfernung der Neoplasie, um signifikante neurologische Defizite zu vermeiden.
Studien haben gezeigt, dass eine unmittelbar nach der Diagnose durchgeführte Strahlentherapie die Zeit verlängert hat, in der der Patient vor dem Wiederauftreten des Tumors krankheitsfrei ist, verglichen mit der Situation, in der sich der Verlauf der Strahlentherapie bis zum Zeitpunkt des Fortschreitens verzögert. Derzeit gibt es jedoch keinen Konsens, dass eine Strahlentherapie kurz nach der Diagnose das „Gesamtüberleben“ des Patienten verbessert.
Bei Patienten mit schwächeren oder keinen Symptomen oder mit mit Antikonvulsiva kontrollierbaren epileptischen Anfällen ist es möglich, die Strahlentherapie hinauszuzögern, bis das Tumorwachstum eine kritische Phase erreicht. Häufig besteht der Wunsch darin, das Risiko einer durch Strahlentherapie selbst verursachten neurologischen Schädigung zu verringern.
Zwei prospektive randomisierte klinische Studien zeigten bei einer hochdosierten Strahlentherapie keinen größeren Nutzen als bei einer niedrigdosierten. In der Regel liegt die Gesamtdosierung zwischen 45 und 54 Gray bei einer Fraktionierung von 1,8 bis 2 Gray.
Die Wirkung der adjuvanten Chemotherapie bei Patienten mit niedriggradigen Astrozytomen wird noch untersucht. Vorläufige Ergebnisse einer klinischen Studie, in der die Strahlentherapie allein mit der Strahlentherapie gefolgt von einer Chemotherapie mit Procarbazin, Lomustin und Vincristin (PCV) verglichen wurde, zeigte einen längeren Zeitraum des „krankheitsfreien Überlebens“ bei der Kombination, aber kein verlängertes „Gesamtüberleben“. Aufgrund der mit dem PCV-Protokoll verbundenen Toxizität wird die Verwendung von Temozolomid sowohl als Ersttherapie als auch nach der Genesung empfohlen.
Anaplastische Astrozytome

Als anaplastisches Astrozytom wird ein bösartiger Hirntumor bezeichnet, der durch diffuses Wachstum, erhöhte Zelldichte und Kernteilungsfiguren gekennzeichnet ist. Er entsteht aus einer bestimmten Zellpopulation des zentralen Nervensystems, den Astrozyten. Der Tumor entspricht nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems einem Grad-III-Tumor.
In der Regel weisen Patienten mit anaplastischem Astrozytom epileptische Anfälle, fokale Neurologiesche Ausfälle, Kopfschmerzen und Persönlichkeitsveränderungen auf. Durchschnittlich beträgt das Patientenalter 45 Jahre. Die Magnetresonanztomographie zeigt im Allgemeinen eine massive Läsion mit erhöhtem Kontrastsignal, das aber auch schwächer ausfallen kann. Die Diagnose wird durch die histologische Untersuchung der Läsion durch Biopsie oder chirurgische Resektion gestellt.
Eine schlechtere Prognose kann bei fortgeschrittenem Alter, schlechter körperlicher Verfassung und signifikanten neurologischen Schäden gegeben sein. Im Allgemeinen ist das therapeutische Ergebnis bei einer vollständigen chirurgischen Resektion (Standardbehandlung) ohne Erhöhung neurologischer Defizite besser. Standardmäßig erfolgt eine Strahlentherapie, da sie nachweislich die Überlebenszeit verlängern kann. Die Rolle der Chemotherapie ist umstritten.
Glioblastome

Die sowohl häufigsten als auch bösartigsten Gliazelltumoren sind Glioblastome. Sie bestehen aus einer heterogenen Masse schlecht differenzierter Astrozytomzellen hauptsächlich bei Erwachsenen. Normalerweise treten sie in den Gehirnhälften auf, seltener am Hirnstamm oder Rückenmark. Außer in sehr seltenen Fällen dehnen sie sich wie alle Hirntumoren nicht über die Strukturen des Zentralnervensystems hinaus aus.
Das Glioblastom kann sich aus einem diffusen (II. Grades) oder einem anaplastischen Astrozytom (III. Grades) entwickeln. In letzterem Fall wird es als sekundär bezeichnet. Tritt es jedoch ohne Vorstadien oder ohne Anzeichen einer früheren Malignität auf, bezeichnet man es als primär. Glioblastome werden mittels Operationen, Bestrahlung und Chemotherapie behandelt. Sie sind schwer zu heilen und es gibt nur wenige Überlebensfälle von über drei Jahren.
Oligodendrogliome
Das Oligodendrogliom ist ein ungewöhnlicher Gehirntumor der Gliazellen, der aus Oligodendrozyten entsteht. Es tritt hauptsächlich bei Erwachsenen zwischen 40 und 45 Jahren vorzugsweise in der Großhirnrinde und der Weißen Substanz der Gehirnhälften auf.
Oligodendrogliome sind relativ selten, weniger als etwa 5 Prozent aller primären Hirntumoren und nicht mehr als etwa 10 bis 15 Prozent aller Gliome. Diese Tumoren sind in niedriggradige und anaplastische Läsionen unterteilt. Das anaplastisches Oligodendrogliom ist durch erhöhte Zelldichte, Mitose, Endothelproliferation und Kernpolymorphismus sowie Nekrosen gekennzeichnet.
Niedriggradige Oligodendrogliome und Oligoastrozytome
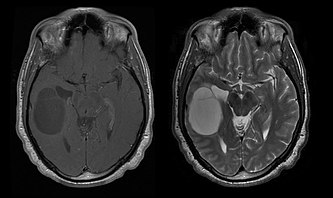
Das mediane Überleben für Patienten mit reinem Oligodendrogliom beträgt etwa 10 Jahre, mit Oligoastrozytom ungefähr 8 Jahre. Die Verlängerung gegenüber reinen Astrozytomen liegt an einer Deletion oder Translokation des 1p/19q-Paares im Tumor.
Das Durchschnittsalter der Patienten bei Diagnose beträgt 35 Jahre. Typische Symptome sind epileptische Anfälle, aber auch fokale neurologische Ausfälle, Persönlichkeitsveränderungen oder andere Symptome des Hirndrucks, wie Kopfschmerzen und Erbrechen, können berichtet werden. Diese Tumoren sind normalerweise im Computertomogramm nicht sichtbar, daher ist die Kernspintomographie die Methode der Wahl für die diagnostische Bildgebung. Auf dem -Kernspintomogramm sind sie als erhöhte Signalintensität erkennbar. Auf -Bildern hingegen kann das Signal gedämpft und die Kontrastverstärkung nur gelegentlich erkannt werden. Eventuell fehlt ein Verkalkungssignal.
Die Entwicklung dieser Tumoren ist träger als die niedriggradiger Astrozytome, und in der Literatur gibt es keine Übereinstimmung hinsichtlich der optimalen Behandlung. Die Erstbehandlung beinhaltet eine Kontrolle der Symptome mit Antikonvulsiva, Strahlen-, Chemotherapie oder einer Kombination aus letzteren beiden. Bei Rückfällen spielen die Chirurgie, Strahlen- und Chemotherapie eine wichtige Rolle. Resektionen können die Symptome lindern. Auf Temozolomid zeigten 50 Prozent der Patienten bei einem Rückfall nach einer Strahlentherapie eine positive Reaktion.
Anaplastische Oligodendrogliome und Oligoastrozytome
Anaplastische Oligodendrogliome weisen typische Symptome auf, die sich aus dem Masseneffekt und epileptischen Anfällen ergeben. Trotz ihrer Chemosensitivität beträgt das mediane Überleben nur 3 bis 5 Jahre. Die Behandlung beinhaltet die größtmögliche Exzision, gefolgt von einer Strahlentherapie. In Bezug auf eine Chemotherapie sollte beachtet werden, dass in zwei kürzlich durchgeführten klinischen Phase-III-Studien die Ergebnisse einer Strahlentherapie mit denen einer kombinierten Strahlen- und Procarbazin, Lomustin, Vincristin-Chemotherapie verglichen. Obwohl die Überlebensdauer ohne relevante Symptome bei der kombinierten Therapie länger war, war das Gesamtüberleben bei beiden Therapien gleich. Patienten mit 1p/19q-Deletion erzielten die besten Therapieresultate, Patienten ohne 1p/19q-Deletion konnten ihre Ergebnisse mittels einer PCV-Chemotherapie verbessern.
Prospektive klinische Studien haben gezeigt, dass etwa 50 bis 70 Prozent der Patienten mit rezidivierendem anaplastischem Oligodendrogliom nach einer Strahlentherapie positiv auf eine Chemotherapie mit PCV oder Temozolomid ansprechen. Obwohl keine überlegene Wirksamkeit der Temozolomid- und PCV-Therapie festgestellt wurde, deutet das Fehlen einer kumulativen Myelosuppression mit Temozolomid auf die Verwendung zu Beginn der Rückfallbehandlung hin.
Ependymome

Das Ependymom ist eine Neoplasie, die sich aus Ependymzellen entwickelt, die die Hirnventrikel, den Plexus choroideus, das Filum terminale und den zentralen Kanal des Rückenmarks auskleiden. Ependymzellen sind auch im Gehirnparenchym infolge der embryonalen Migration von periventrikulären Bereichen zur Großhirnrinde vorhanden.
Diese ziemlich seltenen Tumoren können in jedem Alter auftreten, weisen jedoch zwei charakteristische Peaks auf, von 0 bis 10 und von 40 bis 50 Jahren. Intrakranielle Verletzungen, die normalerweise in der hinteren Schädelgrube auftreten, sind in der ersten Altersgruppe häufiger, Wirbelsäulen Verletzungen dagegen in der zweiten.
Ependymome sind in niedriggradige Läsionen (I. und II. Grades auf der WHO-Skala) und anaplastische Läsionen (III. Grades) unterteilt. I. Grades sind insbesondere Subependymome und myxopapilläre Ependymome, III. Grades das anaplastische Ependymom. Patienten mit niedriggradigen Ependymomen in der Wirbelsäule, die vollständig entfernt werden können, werden danach nicht mehr einer Strahlentherapie unterzogen. Die Rolle der postoperativen Strahlentherapie bei niedriggradigen intrakraniellen Ependymomen ist umstritten, bei anaplastischen oder niedriggradigen Tumoren, die nicht vollständig entfernt werden können, ist eine strahlentherapeutische Behandlung normalerweise angezeigt.
Klinische Studien haben erbracht, dass Ependymome auf Chemotherapien ansprechen, insbesondere auf auf Platin basierende. Der Nutzen bei auf Platin basierenden Chemotherapien beträgt 67 Prozent, bei auf Nitrosoharnstoffen hingegen 25 Prozent. Die Prognosen bei Ependymomen II. Grades liegen bei einem 6-jährigen krankheitsfreien Überleben von 68 Prozent und bei einem Gesamtüberleben von 87 Prozent. Bei anaplastischen Ependymomen fallen diese Werte auf 29 Prozent bzw. auf 37 Prozent.
Nichtgliale Tumoren
Medulloblastome
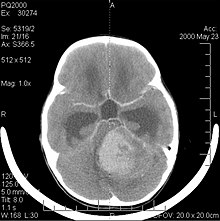
Das Medulloblastom ist der häufigste bösartige Hirntumor im Kindesalter. Die höchste Inzidenz tritt bei Kindern zwischen 2 und 7 Jahren auf. Das größte Krankheitsrisiko besteht weiterhin im Kindesalter, da ein Medulloblastom bei Menschen über 21 Jahre sehr selten ist.
Dieser Tumor ist typisch für die hintere Schädelgrube, wo er in beiden Hemisphären des Kleinhirns oder im Kleinhirnwurm lokalisiert ist. Da er invasiv ist und schnell wächst, breitet er sich normalerweise über den Liquor auf andere Teile des Zentralnervensystems (ZNS) aus und kann den Boden des nahegelegenen vierten Ventrikels und die Hirnhäute infiltrieren. Seltener kann es zu zusätzlichen ZNS-Metastasen kommen. Bei Auftreten der Malignität zählen Gleichgewichtsverlust, mangelnde Koordination, Diplopie, Dysarthrie und aufgrund der Beteiligung des vierten Ventrikels, bei dem häufig ein obstruktiver Hydrozephalus auftritt, Kopfschmerzen, Übelkeit und Erbrechen sowie ein instabiler Gang zu den Symptomen.
Das Kernspintomogramm zeigt normalerweise eine massive Kontrastverstärkungsläsion, an der das Kleinhirn beteiligt ist. Wie oben erwähnt, hat das Medulloblastom eine hohe Neigung, die Leptomeninges lokal zu infiltrieren sowie sich durch den Subarachnoidalraum auszubreiten, und bezieht dabei die Ventrikel, die zerebrale Konvexität und die leptomeningealen Oberflächen der Wirbelsäule mit ein. Folglich ist es notwendig, die gesamte kraniospinale Achse in Resonanz zu bringen.
Die Chirurgie hat die Aufgabe, so viel wie möglich von der durch die Läsion dargestellten Masse zu entfernen. Tatsächlich ergeben postoperative Tumorreste eine schlechtere Prognose. Ein Vorbote einer ungünstigen Prognose ist auch das Vorhandensein von Tumorzellen in der Cerebrospinalflüssigkeit oder der Resonanznachweis von leptomeningealen Metastasen. Eine Operation allein ist normalerweise nicht kurativ. In einigen Fällen kann jedoch eine therapeutische Bestrahlung der kraniospinalen Achse, die sich auf die primäre Tumorstelle konzentriert, die Folge sein. Das Hinzufügen einer Chemotherapie nach einer Strahlentherapie erhöht die Heilungsrate. Arzneimittel auf Platinbasis (Cisplatin oder Carboplatin), Etoposid und ein Alkylierungsmittel (Cyclophosphamid oder Lomustin) werden zusammen mit Vincristin verwendet. Bei entsprechender Behandlung liegen die Fälle eines langen Überlebens von mehr als 3 Jahren bei Medulloblastom-Patienten zwischen 60 und 80 Prozent.
Meningeome

Meningeome sind die häufigsten intrakraniellen extrinsischen oder extraaxialen Hirntumoren, die aus den Zellen der Arachnoidea entstehen, der Membran, die das Gehirn und das Rückenmark bedeckt. Die Inzidenz dieser Neoplasie beträgt etwa 2 Fälle pro Jahr pro 100.000 Einwohner. Sie treten häufiger bei Frauen im sechsten und siebten Lebensjahrzehnt auf. Ihre Häufigkeit ist bei Patienten mit Typ-2-Neurofibromatose höher. Der Verlust des Chromosoms 22 ist charakteristisch für Meningeome, obwohl die prognostische Bedeutung dieses Befundes noch unklar ist.
Patienten mit Meningeom können Symptome aufweisen, die für eine massive Schädelläsion typisch sind, einschließlich Anfällen und fokalen neurologischen Defiziten. Da Meningeom auch asymptomatisch sein können, werden sie manchmal manchmal bei Computer- und Kernspintomographien aus anderen Gründen entdeckt. Dieser Resonanztumor hat ein charakteristisches Erscheinungsbild, das normalerweise aus einer gleichmäßigen Kontrastverstärkung entlang der Dura mit klarer Trennung vom Gehirnparenchym besteht. Ein weiteres Merkmal, obwohl nicht in allen Fällen vorhanden, ist der sogenannte „Duralschwanz“, der durch eine Verstärkung dargestellt wird, die über die Läsion hinausgeht und den Verankerungspunkt in der Dura anzeigt.
Viele zufällig entdeckte Meningeome müssen zum Zeitpunkt der Erstdiagnose nicht behandelt werden. Wenn beim Patienten ein signifikanter Masseneffekt festgestellt wird, unabhängig davon, ob Symptome vorliegen oder nicht, ist die Behandlung der Wahl normalerweise eine vollständige Resektion. In einer Studio der Mayo-Kliniken, in der die Tumorkontrollraten nach chirurgischer Resektion und Radiochirurgie bei Patienten mit kleinem bis mittlerem intrakraniellem Meningeom und ohne Masseneffektsymptome verglichen wurden, führte die Radiochirurgie zu einer besseren Kontrolle (98 gegenüber 88 Prozent) und mit weniger Komplikationen (10 gegenüber 22 Prozent) im Vergleich zur chirurgischen Entfernung.
Primäre ZNS-Lymphome

Primäre Lymphome des Zentralnervensystems machen etwa 2 Prozent bis 3 Prozent aller Hirntumoren von Patienten mit einem normalen Immunsystem aus. Sie treten häufiger bei Männern im Alter von 55 bis 60 Jahren auf. Fast die Hälfte aller Lymphome tritt bei Patienten über 60 Jahren und etwa ein Viertel bei Patienten über 70 Jahren auf. Die Inzidenz scheint mit dem Alter zuzunehmen, der Grund jedoch ist noch ungeklärt. Einem höheren Risiko für ein ZNS-Lymphom ausgesetzt sind Patienten mit einem geschwächten Immunsystem, damit diejenigen, die sich einer Organtransplantation unterzogen haben, einen angeborenen Immundefekt oder eine Autoimmunerkrankung haben oder mit dem Humanen Immundefizienz-Virus infiziert sind. Mit dem HI-Virus assoziierte Hirnlymphome sind mit dem Epstein-Barr-Virus verbunden, insbesondere bei Patienten mit CD4-Lymphozytenanzahl unter 500 Zellen pro Kubikmillimeter im Blut. Die meisten ZNS-Lymphome sind vom Typ her diffuse großzellige B-Zell-Lymphome.
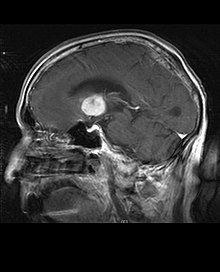
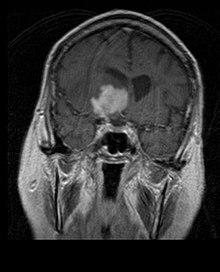
Die Patienten leiden an einer Vielzahl charakteristischer Symptome einer fokalen oder multifokalen massiven Läsion. Das Kernspintomogramm zeigt normalerweise Tumoren mit homogener Kontrastverstärkung innerhalb der tiefen periventrikulären weißen Substanz. Multifokalität und inhomogene Verstärkung sind typisch für Patienten mit geschwächtem Immunsystem. Extrem wichtig ist die Analyse des ZNS-Lymphoms bei der Differentialdiagnose von Hirnneoplasien. Es sollte beachtet werden, dass die Verabreichung von Kortikosteroiden zum vollständigen Verschwinden der Verstärkung führen kann, was Diagnose der Läsionen erschwert. Wenn das ZNS-Lymphom in der Differentialdiagnose berücksichtigt werden soll, sollten folglich Kortikosteroide vermieden werden, es sei denn, der Masseneffekt verursacht ein ernstes und unmittelbares Problem beim Patienten.
Entscheidend ist die Biopsie der vermuteten Läsion. Im Gegensatz zu systemischen großzelligen B-Zell-Lymphomen, bei denen sowohl Chemotherapie als auch Strahlentherapie wirksam sind und die Behandlung lokalisierter Läsionen kurativ ist, spricht das Lymphom des Zentralnervensystems typischerweise auf die Ersttherapie an, tritt dann jedoch erneut auf. Wie beim systemischen Lymphom beschränkt sich die Rolle der Operation in erster Linie darauf, geeignete Gewebeproben für die Diagnose zu erhalten.
Früher wurde das gesamte Gehirn (panenzephal) einer Strahlentherapie unterzogen. Dabei beträgt das mediane Überleben selbst bei lokalisierten Läsionen etwa 12 Monate. Das Wiederauftreten betrifft normalerweise den Ort der vorherigen Verletzung sowie andere Regionen. Die Reaktionen auf eine Chemotherapie sind vielversprechender. Klinische Studien, in denen hochdosiertes Methotrexat allein als erste Behandlung angewendet und die Strahlentherapie auf den Zeitpunkt des Rückfalls oder Fortschreitens verschoben wurde, zeigten ein besseres Gesamtüberleben als die Strahlentherapie allein. Noch effektiver war die Kombination von Methotrexat, Vincristin, Procarbazin, intrathekalem Methotrexat, Cytarabin und panenzephaler Strahlentherapie und Cytarabin bzw. die Anwendung einer intraarteriellen Chemotherapie mit intraarteriell verabreichtem Methotrexat, intravenös injiziertem Cyclophosphamid und Etoposid nach Modifikation der Blut-Hirn-Schranke mit Mannit. Das mediane Überleben in Methotrexat-Therapien war mit 24 bis 40 Monaten viel höher als bei alleiniger Strahlentherapie (Bereich 24 bis 40 Monate). In einigen Fällen wird die Strahlentherapie nur bei Rückfällen angewendet, wenn bei einer Chemotherapie eine anfängliche Regression auftritt. Fälle von langem Überleben wurden auch ohne Strahlentherapie berichtet.
Die panenzephale Strahlentherapie ist mit einem hohen Risiko verbunden, an Demenz oder Leukenzephalopathie zu erkranken. Dieses Risiko könnte durch die Entwicklung wirksamer Tumorkontrollstrategien verringert werden, die eine panenzephale Strahlentherapie vermeiden. Die anfängliche Therapie für Patienten mit geschwächtem Immunsystem besteht darin, die Ursachen der Immunsuppression zu verringern. Die Prognose für diese Patienten ist normalerweise schlechter als die für Patienten, die ein normales Immunsystem vorweisen können. Aufgrund von begleitenden Tumorinfektionen und einer im Allgemeinen suboptimalen körperlichen Verfassung kann bei diesen immunsupprimierten Patienten häufig keine Chemotherapie durchgeführt werden. Wie bei anderen Hirntumoren hängt auch hier das Ansprechen auf die Behandlungen vom Alter und der körperlichen Verfassung ab.
Metastasierende Tumoren des Zentralnervensystems
Hirnmetastasen
Hirnmetastasen sind die häufigsten intrakraniellen Neoplasien bei Erwachsenen, die zehnmal häufiger vorkommen als primäre Hirntumoren. Sie treten bei 20 bis 40 Prozent der krebskranken Erwachsenen auf und sind hauptsächlich mit Lungen- und Brustkrebs sowie Melanomen assoziiert. Diese Läsionen sind das Ergebnis der Ausbreitung von Krebszellen durch den Blutkreislauf und treten am häufigsten an der Verbindung der grauen mit der weißen Substanz auf, wo sich der Querschnitt der Blutgefäße ändert und damit Tumorzellembolien eingeschlossen werden. 80 Prozent der Läsionen treten in den Gehirnhälften auf, 15 Prozent im Kleinhirn und 5 Prozent im Hirnstamm. Ungefähr 80 Prozent der Patienten haben eine Anamnese von systemischem Krebs und 70 Prozent haben multiple Hirnmetastasen.

Bei Diagnose und Behandlung dieser Läsionen wurden in jüngster Zeit erhebliche Fortschritte erzielt, wodurch das Überleben und die Kontrolle der Symptomatik verbessert wurden. Das Auftreten von Anzeichen und Symptomen ähnelt denen anderer massiver Läsionen im Gehirn. Das Diagnoseverfahren der Wahl ist die Kernspintomographie unter Verwendung von Kontrastmitteln.
Die Literatur zeigt äquivalente Ergebnisse für Chirurgie und Radiochirurgie. Letzteres scheint bequemer, effektiver und sicherer für kleine Läsionen oder in Regionen zu sein, die für eine Operation nicht zugänglich sind. Die Radiochirurgie ist eine sinnvolle Alternative für Patienten, die aus medizinischen Gründen nicht operiert werden können. Die Operation ist jedoch eindeutig die optimale Methode, um Gewebe für die Diagnose zu erhalten und die Läsionen zu entfernen, die einen Masseneffekt verursachen. Daher sollten Radiochirurgie und Chirurgie besser als zwei komplementäre, aber unterschiedliche Methoden betrachtet werden, die je nach der unterschiedlichen Situation des Patienten angewendet werden. Für fast 50 Prozent der Patienten mit einem oder zwei Hirnmetastasen kommt eine chirurgische Entfernung aufgrund der Unzugänglichkeit der Läsionen, der Ausdehnung der systemischen Erkrankung oder anderer Faktoren nicht in Frage. Diesen und anderen Patienten mit multiplen Metastasen wird normalerweise eine panenzephale Strahlentherapie als Standardbehandlung angeboten. Tatsächlich erreichen bis zu fast 50 Prozent von ihnen mit dieser Therapie eine Verbesserung der neurologischen Symptome und 50 bis 70 Prozent eine erkennbare Reaktion. Bei Hirnmetastasen wird die Chemotherapie selten primär angewendet.
Bei den meisten Patienten mit Hirnmetastasen beträgt das mediane Überleben nur vier bis sechs Monate nach einer panenzephalen Strahlentherapie. Patienten unter 60 Jahren mit einzelnen Läsionen und einer kontrollierten systemischen Erkrankung können jedoch ein längeres Überleben erzielen, da sie einen aggressiveren Behandlungsansatz vertragen können.
Hirnhautmetastasen

Bei etwa 5 Prozent der Tumorpatienten können Metastasen der weichen Hirnhäute (Leptomeninges encephali) diagnostiziert werden. Am häufigsten treten sie bei Melanomen, Brust- und Lungenkrebs infolge der Ausbreitung von Tumorzellen durch den Blutkreislauf auf. Die malignen Zellen werden dann im Allgemeinen über den Liquor cerebrospinalis, umgangssprachlich auch Gehirnwasser genannt, im gesamten zentralen Nervensystem (ZNS) verbreitet.
Eine oder mehrere der folgenden Anzeichen und Symptome können unter anderem durch Hirnhautmetastasen hervorgerufen werden:
- lokale Nervenschädigungen wie Hirnnervlähmungen, motorische Schwäche und Radikulopathien, Parästhesien und Schmerzen,
- direkte Invasion des Gehirns oder Wirbelsäulengewebes,
- Störung der Blutgefäße in Gehirn und Wirbelsäule mit fokalen neurologischen Defiziten und/oder Anfällen,
- Behinderungen des normalen Flusses des Liquor cerebrospinalis mit Kopfschmerzen und erhöhtem Hirndruck,
- Störungen der normalen Gehirnfunktion wie Enzephalopathie und/oder
- perivaskuläre Infiltration durch Tumorzellen mit daraus resultierender Ischämie und Apoplexiesymptomen.
Die Diagnose kann durch Untersuchung des Liquor cerebrospinalis oder Kernspintomographie des Gehirns und des Rückenmarks gestellt werden. Das Vorhandensein maligner Zellen kann so bei 50 Prozent der Patienten festgestellt werden. Bei mindestens 10 Prozent der Patienten mit leptomeningealer Beteiligung bleibt die zytologische Untersuchung negativ. Durch Erhöhung der Anzahl der Lumbalpunktionen bis auf sechs und der Menge des entfernten Flüssigkeitsvolumens auf 10 Milliliter pro Punktion kann die Möglichkeit einer positiven Diagnose erhöht werden. In der Cerebrospinalflüssigkeit ist die Konzentration von Proteinen normalerweise hoch, die von Glucose kann bei Vorhandensein von Pleozytose niedrig sein. Die radiologische Studie kann einen Hydrozephalus ohne massive Läsion oder diffuse Verstärkung der Leptomeninges zeigen.
Ohne Therapie beträgt das mediane Überleben 4 bis 6 Wochen, wobei der Tod auf eine fortschreitende neurologische Verschlechterung zurückzuführen ist. Leptomeningeale Metastasen sind häufig eine Manifestation des Endstadiums der Hauptkrankheit, und eine symptomatische Therapie kann die am besten geeignete Lösung sein. Kortikosteroide und Analgetika bieten vorübergehende Linderung. Patienten mit minimaler systemischer Erkrankung und akzeptabler allgemeiner körperlicher Verfassung kann eine Behandlung angeboten werden, um die Symptome zu lindern und das Überleben zu verlängern.
Das mediane Überleben kann durch Strahlentherapie auf symptomatische Stellen und voluminösere erkrankte Bereiche, die mittels Röntgendiagnostik identifiziert wurden, sowie durch intrathekale Therapie mit Methotrexat, Cytarabin und Thiotepa, durchgeführt mit Lumbalpunktion oder Ommaya-Katheter, von 3 auf 6 Monate erhöht werden.
Die Hauptkomplikation der intrathekalen Therapie auf Methotrexatbasis ist eine nekrotisierende Leukoenzephalopathie, die sich nach monatelanger Therapie bei den wenigen Patienten entwickeln kann, die ein längeres Überleben genießen können. Diese verheerende toxische Wirkung tritt besonders häufig bei Patienten auf, die zuvor oder gleichzeitig eine Strahlentherapie mit intrathekaler Methotrexat-Therapie erhalten haben.
Schmerzen und Sterbebegleitung
Palliative Care ist eine besondere Form der Pflege zur Verbesserung der Lebensqualität von Patienten, die an einer schweren oder lebensbedrohlichen Krankheit wie Krebs leiden. Ziel der Palliativmedizin ist nicht die Heilung, sondern die möglichst frühzeitige Vorbeugung oder Behandlung der Symptome und Nebenwirkungen der Krankheit und ihrer Behandlung sowie der damit verbundenen psychischen, sozialen und spirituellen Probleme. Palliativpflege wird auch als Komfortpflege, unterstützende Pflege und Symptommanagement bezeichnet.
Palliativpflege wird während der gesamten Krebserfahrung eines Patienten geleistet. Sie beginnt in der Regel mit der Diagnose und setzt sich über die Behandlung, Nachsorge und das Lebensende fort.
Weblinks
- www.neuroonkologie.de – Neuroonkologische Arbeitsgemeinschaft (NOA)
Literatur
- Jan C. Buckner et al.: Central Nervous System Tumors. In: Mayo Clinic Proceedings. Jg. 82, Nr. 10, 2007, S. 1271–1286.
- Lisa M. DeAngelis et al.: Intracranial Tumors. Diagnosis and Treatment. Dunitz, London 2002, ISBN 1-901865-37-1.
- D. N. Louis et al. (Hrsg.): WHO Classification of Tumours of the Central Nervous System. 4. Auflage. Genf 2007, ISBN 978-92-832-2430-3.
- Richard Pazdur et al.: Cancer management. A multidisciplinary approach. Medical, surgical, & radiation oncology. UBM Medica, Norwalk 2010, ISBN 978-0-615-41824-7.
- Jerome B. Posner: Neurologic Complications of Cancer. Davis, Philadelphia 1995, ISBN 0-8036-0006-2.
- Rüdiger Schenk: Neuroonkologische Therapiekonzepte zur Behandlung von Astrozytomen höheren Malignitätsgrades und Rezidivlokalisation. Regensburg 2019.
- Uwe Schlegel et al. (Hrsg.): Neuroonkologie. 2. erw. Auflage. Thieme, Stuttgart 2003, ISBN 3-13-109062-6.
- Jörg-Christian Tonn et al.: Oncology of CNS Tumors. 2. Auflage. Springer, Berlin 2010, ISBN 978-3-642-02873-1.