
| Klassifikation nach ICD-10 | |
|---|---|
| Q16 | Angeborene Fehlbildungen des Ohres, die eine Beeinträchtigung des Hörvermögens verursachen |
| Q17 | Sonstige angeborene Fehlbildungen des Ohres |
| ICD-10 online (WHO-Version 2019) | |
Unter dem Begriff Ohrmuschelfehlbildung werden sowohl funktional nicht beeinträchtigende Anomalien wie abstehende Ohren als auch ausgeprägtere Ohrmuschelveränderungen wie Blumenkohlohren bis hin zum völligen Fehlen der Ohrmuschel zusammengefasst. Als Teil des Gesichts gehört die Ohrmuschel zu einer hervorgehobenen Körperregion, deren Auffälligkeit für das Familienleben, den Beruf und die soziale Integration Bedeutung haben kann.
Epidemiologie/Ätiologie
Eine höhergradige Mikrotie tritt in der westlichen Bevölkerung in 0,76 bis 2,35 Fällen pro 10.000 Geburten auf. Legt man die aktuelle Geburtenrate zugrunde, kommen in Deutschland etwa 100 bis 150 neue Fälle pro Jahr hinzu.
In der Regel tritt die Fehlbildung isoliert auf. In 20 bis 30 % der Fälle tritt sie kombiniert mit anderen Fehlbildungen wie Gesichtshypoplasie, Lippen-Kiefer-Gaumenspalten, inneren Fehlbildungen, kognitiver Beeinträchtigung oder im Rahmen von genetisch bedingten Syndromen, z. B. dem Aurikulo-kondylären Syndrom oder dem Mengel-Konigsmark-Berlin-McKusick-Syndrom auf. Die häufigsten mit Mikrotie assoziierten Syndrome sind das Goldenhar-Syndrom und das Franceschetti-Syndrom.
Als Ursachen werden hämorrhagische Ereignisse in der Frühschwangerschaft, Schwangerschaftsdiabetes und genetische Ursachen diskutiert. Weitergehende Untersuchungen hierzu stehen aber noch aus. Familiäre Häufungen werden in Einzelfällen beobachtet, hier liegt meist ein autosomal dominanter Erbgang mit variabler Penetranz zugrunde. Die große Mehrheit tritt jedoch sporadisch auf. Die meisten Autoren sehen in der isolierten Mikrotie eine Minimalvariante der Hemifazialen Mikrosomie. Die Genese ist dabei vermutlich multifaktoriell mit einer zugrunde liegenden genetischen Wahrscheinlichkeit, auslösend wirken äußere Faktoren wie z. B. Blutungen.
Einteilung
Bei der Einteilung der Ohrmuschelfehlbildungen werden drei Grade unterschieden.
- Bei der Dysplasie °1 sind die meisten anatomischen Strukturen der Ohrmuschel vorhanden. Hierzu zählen u. a. abstehende Ohren (Apostasis otum), Tassenohr °I und Tassenohr °II.
- Bei der Dysplasie °2 sind nur einige Strukturen der normalen Ohrmuschel vorhanden. Hierzu zählen u. a. Tassenohr °III und das Miniohr.
- Bei der Dysplasie °3 sind normale Ohrmuschelstrukturen praktisch nicht vorhanden, es existieren lediglich Rudimente. Hierzu zählen u. a. Mikrotie °III, Anotie und die Dystopie.
Diagnostik
Menschen mit Mikrotie müssen frühzeitig interdisziplinär beraten und behandelt werden. Dabei sollten HNO-Ärzte, Kinder- und Jugendärzte, Phoniater und Pädaudiologen sowie gegebenenfalls Humangenetiker und Mund-Kiefer-Gesichts-Chirurgen beteiligt sein. Anamnestisch sollte eine familiäre Häufung und schädigende Einflüsse in der frühen Schwangerschaft erfragt werden. Bei familiärer Häufung empfiehlt sich die Vorstellung in der Humangenetik, um die Auftrittswahrscheinlichkeit bei weiteren Kindern abschätzen zu können. Eine Computertomographie zur Beurteilung der Mittelohrstrukturen wird häufig im 10. Lebensjahr, in der Regel während des ersten stationären Aufenthaltes, durchgeführt.
Sprachentwicklung
Nach heutiger Lehrmeinung soll die Lautsprachentwicklung bei einem einseitigen Schallleitungsblock und normalem Gehör auf der gesunden Seite ungestört verlaufen. Deshalb ist die nicht betroffene Seite regelmäßig fachärztlich zu überprüfen, und Schallleitungs- oder Schallempfindungsschwerhörigkeiten müssen zügig und konsequent behandelt werden. Kinder mit beidseitigen Fehlbildungen bzw. beidseitigem Schallleitungsblock benötigen rasch nach der Geburt eine akustische Verstärkung. Dazu werden die Kinder zunächst mit einem Stirnband-Knochenleitungshörgerät versorgt, im 4. Lebensjahr können dann knochenverankerte Geräte (BAHA) zum Einsatz kommen. Bei der Implantation der Knochenanker ist darauf zu achten, dass diese nicht zu nahe am Ohrmuschelrudiment eingebracht werden, um eine spätere ästhetische Rekonstruktion nicht durch Narben zu erschweren.
Therapie
Einige Ohrmuscheldysplasien °I (Stahlsohr, Tassenohr, fehlende Helixausformung, Satyrohr, abstehende Ohrmuschel, eingerolltes Ohr (Tanzer Typ I)) sollten bereits bei Neugeborenen nicht invasiv behandelt werden. Der Knorpel des Neugeborenen ist in den ersten Lebenswochen formbar und kann mit einem Ohrmuschelformer aus Silikon und individuell anpassbaren Traktoren für das Neugeborene schmerzfrei geformt werden. Diese Behandlungsmethode erzielt den größten Erfolg, wenn sie zwischen dem fünften und siebten Lebenstag des Säuglings begonnen wird. Je älter das Kind wird, desto steifer wird der Knorpel des Ohres und lässt sich allenfalls ein wenig formen. Amerikanische Studien belegen eine Erfolgsquote von 90 % wenn die Behandlung in den ersten Lebenstagen begonnen wird.
Eine operative Korrektur der Ohrmuschel setzt mindestens ein Alter von vier bis fünf Jahren voraus, um der körperlichen Reife für eine invasive Operation zu entsprechen.
Ohrmuscheldysplasien °I können durch Bearbeitung vorhandenen Gewebes behandelt werden, zum Beispiel durch Otopexie. Ohrmuscheldysplasien höheren Grades erfordern den Einsatz von Verschiebeplastiken, Transplantaten oder Implantaten wie körpereigenem Knorpel oder Kunststoff. Anzustreben ist eine möglichst natürliche Rekonstruktion, die der Form der Gegenseite entspricht.
Der chirurgische Aufbau eines fehlgebildeten Ohres mit körpereigenem Knorpel erfolgt in zwei bis drei Schritten im Abstand von drei Monaten. Körpereigener Rippenknorpel ist das Material, mit dem derzeit weltweit die größten Erfahrungen vorliegen.
- Während des ersten Schrittes wird der Rudimentknorpel entfernt, die Rudimenthaut präpariert, der Rippenknorpel entnommen und daraus das dreidimensionale Ohrmuschelgerüst individuell gefertigt und implantiert.
- Während des zweiten Schrittes wird die Ohrmuschel vom Untergrund abgehoben und die Falte hinter dem Ohr gebildet. Dazu wird der Restknorpel aus dem ersten Operationsschritt als Abstandshalter hinter der Ohrmuschel befestigt. Die Deckung des Hautdefektes erfolgt sowohl durch lokale Hautverschiebung als auch mit einem freien Hauttransplantat.
- Während des dritten Schrittes können erneut Korrekturen der Vorderseite vorgenommen werden. Hierbei steht die Entfernung der Hautüberschüsse aus dem ersten Rekonstruktionsschritt im Vordergrund. Gleichzeitig können einzelne Details der Ohrmuschel weiter herausgearbeitet werden. In einigen Fällen kann dieser Schritt entfallen.
Alternativ ist der chirurgische Aufbau eines fehlgebildeten Ohrs mit Implantaten aus Kunststoff möglich. Die individuell formbaren Kunststoff-Implantate werden mit einem Gewebelappen aus dem Schläfenbereich und freien Hauttransplantaten bedeckt. Die Operation kann häufig in einem Schritt durchgeführt werden, eine Entnahme von Rippenknorpel ist nicht notwendig. Die besten Langzeiterfahrungen liegen mit Implantaten aus porösem Polyethylen vor, die seit den 1980er Jahren zum Einsatz kommen, industriell gefertigt werden und für die chirurgische Verwendung zugelassen sind. Den Anstoß zur Anwendung von Polyethylenimplantaten für die Ohrrekonstruktion hat Alexander Berghaus mit experimentellen und klinisch-wissenschaftlichen Arbeiten gegeben.

Dysplasie °I (Segelohren)
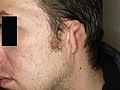
Dysplasie °III (Mikrotie)

