
| Klassifikation nach ICD-10 | |
|---|---|
| D57.0 | Sichelzellenanämie mit Krisen
Hb-SS-Krankheit mit Krisen |
| D57.1 | Sichelzellenanämie ohne Krisen |
| D57.2 | Doppelt heterozygote Sichelzellenkrankheit |
| D57.3 | Sichelzellen-Erbanlage
Heterozygotes Hämoglobin S |
| D57.8 | Sonstige Sichelzellenkrankheiten |
| ICD-10 online (WHO-Version 2019) | |
Die Sichelzellkrankheit (auch Sichelzell(en)anämie) oder Drepanozytose ist eine erbliche Erkrankung der roten Blutkörperchen (Erythrozyten). Sie gehört zur Gruppe der Hämoglobinopathien (Störungen des Hämoglobins) und führt zu einer korpuskulären hämolytischen Anämie. Bei den Betroffenen liegt eine Mutation der β-Kette des Hämoglobins vor. Es können entweder alle β-Ketten betroffen sein (schwere, homozygote Form) oder nur ein Teil (mildere, heterozygote Form).
Die Krankheit tritt vor allem bei Personen aus Subsahara-Afrika und deren Nachfahren, aber auch in Teilen des Mittelmeerraums und des Nahen Ostens bis Indien auf und wurde durch Migration global verbreitet. Sie ist nach wie vor in den Entwicklungsländern mit einer hohen Mortalität verbunden. Die Krankheit wurde 1910 von James Herrick und Ernest E. Irons bei einem Patienten aus der Karibik beschrieben und die Bezeichnung Sichelzellenanämie wurde zuerst von Verne R. Mason 1922 benutzt.
Eigenschaften
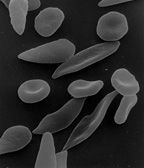

Die Betroffenen bilden ein abnormes Hämoglobin (Sichelzell-Hämoglobin, HbS), das bei Sauerstoffmangel zur Bildung von Fibrillen neigt. Dabei verformen sich die roten Blutzellen durch die enthaltenen Fasern zu sichelförmigen Gebilden, verklumpen miteinander und verstopfen kleine Blutgefäße, wodurch eine Entzündung entsteht. Durch die Verklumpung und Gefäßverstopfung kann es bei der homozygoten Form zu anfallsartigen schmerzhaften, zum Teil lebensbedrohlichen Durchblutungsstörungen (Sichelzellkrisen) kommen, die unter anderem zu venösen Thrombosen führen können. Heterozygot Betroffene, bei denen nur eines der beiden Hämoglobin-Gene verändert ist, sind vor den schweren Verlaufsformen der Malaria geschützt. Dadurch ist das mutierte Hämoglobin-Gen in Malariagebieten relativ verbreitet.
Die Zerstörung roter Blutkörperchen führt zu einer schweren chronischen Blutarmut (hämolytische Anämie). Aufgrund der Neigung des Hämoglobin S zur Polymerisation und der sichelförmigen Deformierung der Erythrozyten kommt es zu Verschlüssen kleiner Arterien mit rezidivierenden Durchblutungsstörungen. Dies führt zu starken Schmerzen und Schäden in multiplen Organsystemen: Gehirn (ischämischer Schlaganfall), Milzinfarkt, Lunge (Lungenentzündung, pulmonale Hypertonie), Auge, Herz- und Nierenversagen, Muskel, Knochen (Osteonekrose) oder Priapismus. Die Lebenserwartung ist vermindert. Eine Glomerulopathie mit Hyperfiltration tritt bei bis zu einem Drittel der Patienten mit homozygotem Phänotyp schon in der Kindheit auf. Schäden im Bereich des Nierenmarks führen zu Papillennekrosen, Verlust der Konzentrationsfähigkeit der Nieren und blutigem Urin (Makrohämaturie). Schäden im Bereich der Nierenkörperchen (Glomeruli) führen zu vermehrter Eiweißausscheidung im Urin (Mikro- und Makroalbuminurie, nephrotisches Syndrom). Bei der feingeweblichen Untersuchung ist die vorherrschende glomeruläre Schädigung die fokal segmentale Glomerulosklerose. Bei bis zu einem Drittel der Patienten kommt es in den ersten Lebensjahrzehnten zum Auftreten einer Proteinurie, in fünf Prozent zu einem terminalen Nierenversagen.
Nur homozygote Träger des Sichelzellgens zeigen diese starke Ausprägung der Krankheit, bei der das gesamte Hämoglobin abnormes Sichelzellhämoglobin (irreguläres Hämoglobin) ist. In heterozygoten Trägern ist nur etwa ein Prozent aller Erythrozyten deformiert. Die Symptome verschlimmern sich erheblich, wenn die Patienten körperlich stark aktiv sind oder sich in großen Höhen befinden. Dies liegt daran, dass sich die Sichelform der Erythrozyten bei niedrigem Sauerstoffpartialdruck bildet, weil unter diesen Bedingungen das Hämoglobin faserig ausfällt (die Löslichkeit von Hämoglobin bei Sichelzellanämie ist 25-fach geringer als die Löslichkeit von normalem Hämoglobin).
Etwa ab dem sechsten Lebensmonat, wenn der Abbau des fetalen Hämoglobins bereits weit fortgeschritten ist, können erstmals Symptome auftreten. Sie äußern sich dann meist in einer sogenannten Sichelzellkrise: Durch äußere Einflüsse wie Anstrengung sinkt der Sauerstoffpartialdruck im Blut, die Sichelzellen werden hämolytisch.
Ursache
Aufgrund einer Punktmutation im HBB-Gen (c.20A>T) auf Chromosom 11 ist bei der Sichelzellenanämie an der Position sechs der β-Globin-Protein-Untereinheit des Hämoglobins die Aminosäure Glutaminsäure durch Valin ersetzt. Die Bezeichnung dieser Variante in offizieller genetischer Nomenklatur lautet HBB-p.E6V. Die betroffenen Erythrozyten verformen sich bei abnehmendem Sauerstoffpartialdruck sichelförmig, verfangen sich leicht in den Kapillaren und lysieren überdies sehr schnell. Durch die Hämolyse werden Hämoglobin, Arginase und freie Sauerstoffradikale freigesetzt. Freies Hämoglobin bindet Stickstoffmonoxid etwa 1000-mal stärker als intrazelluläres, und Arginase verwandelt Stickstoffmonoxid zu Nitrit und Nitrat. Stickstoffmonoxid ist der wichtigste Vasodilatator, und die Konzentrationsabnahme führt zur Gefäßverengung und somit zu Durchblutungsstörungen.
Das Sichelzellenhämoglobin wird als HbS bezeichnet im Gegensatz zum HbA, dem normalen Hämoglobin des erwachsenen Menschen. Heterozygote Träger des Merkmals bilden neben dem HbS auch HbA in ausreichender Menge, um die Funktion der Erythrozyten bei diesen Menschen weitgehend aufrechtzuerhalten.
Am Beispiel der Sichelzellenanämie wurde erstmals der Zusammenhang eines Defekts eines Moleküls mit einer Krankheit nachgewiesen, in einer berühmten Arbeit von Linus Pauling, Harvey Itano und Seymour Jonathan Singer von 1949. Der Unterschied des Hämoglobins beider roter Blutkörperchen zeigte sich in der Gel-Elektrophorese, die Itano durchführte. Die Autoren vermuteten schon, dass Unterschiede in den Aminosäuren vorlagen, was Vernon Ingram 1956 bestätigte, der auch zeigte, dass der Unterschied im Austausch genau einer Aminosäure bestand. Die Vererbungsmuster der Krankheit klärte ebenfalls 1949 James V. Neel (1915–2000).
Vererbung
Sichelzellenanämie ist eine autosomal kodominante Erbkrankheit.
- Das Erbgut eines Gesunden enthält die beiden unvollständig dominanten (auf molekularem Niveau sind A und S kodominant) Allele (AA) für das Hämoglobin A. Seine roten Blutkörperchen sind stets elastisch.
- Ein Überträger (Konduktor) mit dem Genotyp AS (=heterozygot) enthält sowohl das Allel A als auch das mutierte Allel S, welches das veränderte Hämoglobin S verursacht. Seine roten Blutkörperchen enthalten HbA und HbS im Verhältnis 1:1. Unter Normalbedingungen zeigen die roten Blutkörperchen keine Veränderungen, die Krankheit kommt nicht zum Ausbruch. Erst unter sehr starkem Sauerstoffmangel verformen sich die roten Blutkörperchen zu sichelförmigen Gebilden, was die Durchblutung der Organe beeinträchtigt.
- Ein Träger des Genotyps SS (homozygot) stellt nur das veränderte HbS her. Schon unter physiologischem Sauerstoffmangel, wie er z. B. in den Kapillaren sauerstoffverbrauchender Organe herrscht, kommt es zu einer starken Verformung der roten Blutkörperchen. Sie verlieren ihre Elastizität und verhaken leicht miteinander. Dadurch kommt es zu einem Verschluss der Kapillaren. Unter normalen Bedingungen ist das Hämoglobin in den roten Blutkörperchen stets fein verteilt. Bei abnehmendem pH-Wert und Sauerstoffgehalt des Blutes kommt es beim HbS zu einer Verklumpung der Hämoglobinmoleküle zu stäbchenförmigen, kristallinen Gebilden. Dadurch wird der Erythrozyt sichelförmig verformt und verliert seine Elastizität.
Diagnose
Die Diagnose der Sichelzellanämie erfolgt zunächst anamnestisch, wobei die Herkunft und andere Fälle der Erkrankung in der Familie erfragt werden sollten; danach klinisch anhand der gebotenen Symptome und schließlich im Labor, wo in einem Blutbild eine hämolytische Anämie erscheinen kann und bei Untersuchung des Blutes unter dem Mikroskop die typisch geformten Drepanozyten erscheinen - insbesondere, wenn das Blut für 24 h unter Sauerstoffabschluss gelagert wurde (Sichelzelltest). Außerdem kann eine Elektrophorese des Hämoglobins die veränderten Moleküle beweisend identifizieren. Schließlich kann auch der Genabschnitt für das Hämoglobin in einer Restriktionsanalyse untersucht werden und die Punktmutation auf der Ebene der DNA aufzeigen.
Stammbaumanalyse
Sind die Genotypen der Eltern bekannt, kann die Wahrscheinlichkeit berechnet werden, mit der die Sichelzellenanämie bei einem Kind auftritt:
| Genotyp der Eltern |
Genotypen der Kinder |
Vererbungswahrscheinlichkeiten und Phänotyp der Kinder | |
|---|---|---|---|
| AA x AA | AA | 100 % | gesunde Kinder |
| AA × AS | AA | 50 % | Wahrscheinlichkeit, dass ein Kind gesund ist |
| AS | 50 % | Wahrscheinlichkeit, dass ein Kind heterozygoter HbS-Träger wird | |
| AS × AS | AA | 25 % | Wahrscheinlichkeit, dass ein Kind gesund ist |
| AS | 50 % | Wahrscheinlichkeit, dass ein Kind heterozygoter HbS-Träger wird | |
| SS | 25 % | Wahrscheinlichkeit, dass ein Kind von der schwereren, homozygoten Form betroffen ist | |
| AA × SS | AS | 100 % | alle Kinder werden heterozygote HbS-Träger |
| AS × SS | AS | 50 % | Wahrscheinlichkeit, dass ein Kind heterozygoter HbS-Träger wird |
| SS | 50 % | Wahrscheinlichkeit, dass ein Kind von der schwereren, homozygoten Form betroffen ist | |
| SS × SS | SS | 100 % | Die Kinder haben mit Sicherheit die schwerere, homozygote Form |
Sichelzelltest
Das zu untersuchende Blut wird mit EDTA versetzt und über 24 Stunden unter Sauerstoffabschluss gelagert. Durch den eintretenden Sauerstoffmangel in den Erythrozyten entstehen die Sichelformen der Zellen, die im Mikroskop bei 40-facher Vergrößerung gut erkannt werden können. Zusätzlich kann durch Zugabe von Natriummetabisulfit der Effekt der Sichelzellbildung beschleunigt werden.
Elektrophorese

Da erst unter extremem Sauerstoffmangel eine Veränderung der roten Blutkörperchen von Überträgern (Genotyp AS) auftritt, lässt sich durch Untersuchung der roten Blutkörperchen unter dem Mikroskop der Genotyp AA nicht vom Genotyp AS unterscheiden. Dagegen lässt sich mit Hilfe der Hämoglobin-Elektrophorese eindeutig der Genotyp bestimmen: Dazu wird den Probanden Blut entnommen und aufbereitet, bis reines Hämoglobin vorliegt. Im elektrischen Feld wandern die beiden Hämoglobinsorten unterschiedlich weit, da HbS aufgrund seiner geänderten Proteinstruktur ein anderes Wanderungsverhalten zeigt. Auch klinisch relevante Kombinationsformen, wie z. B. aus HbS und Hämoglobin C, die s.g. HbSC-Krankheit oder die Kombination aus HbS und Hämoglobin E (HbE) können mit der Hb-Elektrophorese unterschieden werden.
Molekulargenetik
Die ursächliche Mutation im HBB-Gen (c.20A>T) kann mittels molekularbiologischer Untersuchungsverfahren nachgewiesen werden. Die Sequenzierung des HBB-Gens verdrängt hierbei zunehmend andere Verfahren wie bspw. die Restriktionsanalyse.
Verbreitung
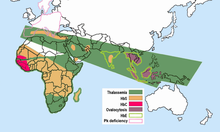
Auffallend ist, dass in Gebieten der Malaria das Sichelzellenallel relativ häufig ist. Daraus wurde geschlussfolgert, dass es gegen Malaria eine Resistenz verleiht, sodass die gesunden Überträger (AS) des Sichelzellenallels in diesen Gebieten einen Selektionsvorteil (den sogenannten Heterozygotenvorteil) gegenüber denen ohne Sichelzellenallel (Genotyp AA), die eher an Malaria sterben, und auch gegenüber den Sichelzellerkrankten (Genotyp SS) haben, die vorzeitig an Sichelzellenanämie sterben. In Afrika gibt es beispielsweise Gegenden, in denen fast ein Drittel der Bevölkerung heterozygot für dieses Merkmal ist. In den anderen Weltgegenden kommt das Sichelzellenallel praktisch nicht vor, da hier dieser Selektionsvorteil aufgrund der fehlenden Malaria nicht wirksam ist. In Deutschland werden jährlich zwischen 500 und 1100 Fälle von Malaria festgestellt. Etwa 85 % der homozygoten Träger stammen aus Afrika.
Verlässliche Daten zur Häufigkeit in Deutschland liegen nicht vor. Die Gesamtzahl der in Deutschland lebenden Patienten mit Sichelzellkrankheit wurde für das Jahr 2017 auf mindestens 2000 geschätzt.
Bedeutung der Sichelzellanämie für Malaria

Möglicherweise besteht ein Selektionsvorteil von heterozygoten Trägern bei Infektionen mit Malaria. Der Malaria-Erreger wird während eines Teiles seines Entwicklungszyklus an oder in den Erythrozyten transportiert. Das Hämoglobin von Menschen mit der heterozygoten Form des HbS führt durch Verminderung der Sauerstoffsättigung des Hämoglobins unter Extrembedingungen zur sichelartigen Verformung der roten Blutzellen, die dann in der Milz abgebaut werden oder verklumpen und danach zugrunde gehen. Eine Hypothese besagt, diejenigen Zellen, die von Plasmodien befallen sind, würden sich auch ohne diese Druckverminderung schon allein durch den Einfluss der Merozoiten beziehungsweise der Trophozoiten verformen, von der Milz als krank erkannt und abgebaut werden.
Eine weitere Hypothese ist die direkte Tötung der Parasiten, denn die Sichelzellen bilden vermehrt Sauerstoffradikale. Es entstehen dabei Superoxidanionen und Wasserstoffperoxid, und beide Verbindungen sind für die Parasiten giftig.
Eine andere Theorie besagt Folgendes: Befallen die Plasmodien, die die Malaria verursachen, Erythrozyten, so setzen die Mikroben nach einiger Zeit Säuren als Abfallprodukte ihres Stoffwechsels frei. Das Hämoglobin gibt nun, von H+-Ionen angeregt, den Sauerstoff ab (Rechtsverschiebung der Sauerstoffbindungskurve). Die Sichelform betrifft aber vor allem die Desoxyform der Erythrozyten. Also werden die befallenen Zellen schnell zu Sichelzellen, und diese werden dann in der Milz samt den Mikroben abgebaut. Daraus erklärt sich die Resistenz der Träger der Sichelzellenanämie gegenüber Malaria (siehe Abbildung).
Die letzte Theorie besagt, dass dabei unter der Bildung von Hämoglobinpolymeren Hämin entsteht, das wiederum zur direkten Tötung der Parasiten führt.
Therapie
Momentan werden Ansätze zur Verstärkung der Genexpression von HbF in Adoleszenten und Erwachsenen untersucht.
Hydroxyurea kann die Bildung von HbF induzieren. Rote Blutkörperchen mit hohem Anteil von HbF bilden keine Sichelzellen, werden daher seltener abgebaut und verursachen seltener Verschlüsse kleiner Gefäße. Eine Behandlung mit Hydroxyurea kann die Häufigkeit von Gefäßverschlüssen vermindern, chronische Organschäden mildern und das Überleben verlängern. Dieses konnte kürzlich auch bei Kindern im Subsahara-Afrika gezeigt werden.
Weiterhin wird ein adoptiver Zelltransfer untersucht.
Am 12. Oktober 2016 wurde eine Therapiemöglichkeit basierend auf einer Veränderung der betroffenen Gene mit der CRISPR/Cas-Methode veröffentlicht. Mit Hilfe der Genschere ersetzten die Forscher die krankmachende Mutation durch die korrekten DNA-Basen. Es wurden zum ersten Mal ausreichend gesunde Blutzellen erzeugt, um Patienten künftig mit dieser Methode heilen zu können, wie die Forscher um Jacob Corn von der University of California, Berkeley, berichteten. Es sei noch zu früh, um von einer praktikablen Lösung zu sprechen, jedoch sei der erste Schritt gemacht um die Ursachen zu bekämpfen, anstatt eine Symptombehandlung durchzuführen.
Weitere Arzneimittel sind der Sauerstoffaffinitäts-Modulator Voxelotor und der monoklonale Antikörper Crizanlizumab.
Literatur
- Fernando Ferreira Costa, Nicola Conran (Hrsg.): Sickle Cell Anemia. From Basic Science to Clinical Practice. Springer 2016
Weblinks
- Ausführlicher praktischer Leitfaden der Uni Bonn
- Sickle Cell Disease (englisch) Webseite der Harvard-Universität zu den Sichelzellkrankheiten
- Sickle Cell Anemia (englisch) Ausführliche Seite von U.S. National Library of Medicine und National Institutes of Health
- Sickle Cell Disease (englisch) Website des Cold Spring Harbor Laboratory mit vielen Animationen und Videos
- IST e.V. – Interessengemeinschaft Sichelzellkrankheit und Thalassämie
- deutschlandfunk.de Wissenschaft im Brennpunkt 16. Dezember 2018, Thomas Reintjes: Experimentelle Therapien für Sichelzellenkranke