

SARS-CoV-2 (Abkürzung für englisch severe acute respiratory syndrome coronavirus type 2‚ Schweres-akutes-Atemwegssyndrom-Coronavirus Typ 2) ist ein Betacoronavirus, das nach dem erstmaligen Nachweis zunächst auch als neuartiges Coronavirus oder nur als Coronavirus bezeichnet wurde. Es ist mit dem Betacoronavirus SARS-CoV-1 verwandt, welches das schwere akute Atemwegssyndrom (SARS) verursacht und die SARS-Pandemie 2002/2003 ausgelöst hat.
SARS-CoV-2 wurde Anfang 2020 als Auslöser der Infektionskrankheit COVID-19 identifiziert, die laut Chinas Regierung erstmals Ende 2019 in der chinesischen Stadt Wuhan als „Lungenkrankheit unbekannter Genese“ in Erscheinung trat. Die Weltgesundheitsorganisation (WHO) stufte COVID-19 am 30. Januar 2020 als „gesundheitliche Notlage von internationaler Tragweite“ ein und klassifizierte das Auftreten der Erkrankung auf Grund der weltweiten Ausbreitung am 11. März 2020 als Pandemie (siehe COVID-19-Pandemie).
Das Genom von SARS-CoV-2 ist über 29,7 kb (29,7 knt) groß und damit eines der umfangreichsten unter den RNA-Viren. Das Virion von SARS-CoV-2 ist zwischen 50 und 140 Nanometern groß und wird in der Regel im nahen menschlichen Kontakt durch Tröpfchen und Aerosole übertragen. Für die weltweite Ausbreitung spielten besonders größere Übertragungsereignisse, sogenannte Superspreading-Events, eine wichtige Rolle.
SARS-CoV-2 hat mittlerweile zahlreiche Varianten mit Mutationen des Spike-Proteins ausgebildet. Klinisch-diagnostische und epidemiologische Erfahrungen sprechen dafür, dass bestimmte Virusvarianten schwerere Krankheitsverläufe verursachen können. Die Mitte 2021 weltweit grassierende Delta-Variante verbreitete sich schneller als zuvor der Wildtyp des Virus und wird leichter von Mensch zu Mensch übertragen. Mit den bisher verfügbaren Impfstoffen geimpfte Personen können das Virus in ähnlichem Maße wie ungeimpfte übertragen – so die WHO im August 2021. Seit Januar 2022 dominiert die Omikron-Variante das Infektionsgeschehen während der COVID-19-Pandemie mit etwa 90 % weltweit.
Entdeckungsgeschichte
Im Dezember 2019 wurden in der Großstadt Wuhan gehäuft schwere Lungenentzündungen unbekannter Ursache festgestellt. Am 30. Dezember 2019 informierte der chinesische Arzt Li Wenliang in einer WeChat-Gruppe seine Kollegen über sieben Patienten, die wegen Verdachts auf Infektion mit dem Betacoronavirus SARS-CoV-1 im Zentralkrankenhaus Wuhan behandelt wurden. Li selbst erkrankte später an COVID-19 und starb daran.
Neben den Coronaviren SARS-CoV-1, SARS-CoV-2 und MERS-CoV zirkulieren vier weitere Viren aus der Familie der Coronaviridae – die zwei Alphacoronaviren HCoV-NL63 und HCoV-229E und die zwei Betacoronaviren HCoV-HKU1 und HCoV-OC43 – beim Menschen, sind weltweit verbreitet und verursachen 10–15 % aller Erkältungskrankheiten. Die von den letztgenannten vier Coronaviren verursachten Infektionen zeigen saisonale Muster und treten meist in den Wintermonaten auf. Es wird angenommen, dass diese vier Viren alle zoonotischen Ursprungs sind. Das Betacoronavirus HCoV-OC43 wurde als potenzieller ätiologischer Erreger der als „Russische Grippe“ bekannten Pandemie vorgeschlagen, der um 1890 weltweit bis zu einer Million Menschen zum Opfer fielen und die erst durch die Spanische Grippe übertroffen wurde, die ab 1918 weit über 25 Millionen Opfer forderte.
Das Chinesische Zentrum für Krankheitskontrolle und -prävention entsandte am 31. Dezember 2019 ein Team in die Stadt. Am selben Tag wurde das China-Büro der WHO durch die chinesischen Behörden offiziell informiert, dass im Dezember 2019 in Wuhan mehrere Personen an schwerer Lungenentzündung erkrankt waren und dass als deren Ursache ein uncharakterisierter Erreger vermutet werde. Bis zum 3. Januar 2020 wurden der WHO insgesamt 44 Erkrankte gemeldet, darunter auch Schwerkranke. Da mehrere Erkrankte auf dem „Großhandelsmarkt für Fische und Meeresfrüchte in Wuhan“ gearbeitet hatten – ein Nassmarkt, auf dem außer Fleischwaren, Fisch und Meeresfrüchten meist noch lebende oder kurz vor dem Verkauf geschlachtete Tiere, auch Reptilien und vielerlei andere Fleischwaren angeboten werden – wurde dort der Ursprung von COVID-19 vermutet. Kurz nach Auftreten der Viruserkrankung im Dezember 2019 hatten 27 (66 %) der ersten 41 Krankenhauspatienten den Nassmarkt im Zentrum Wuhans besucht; 13 der übrigen Betroffenen wurden nicht im Zusammenhang mit einem Marktbesuch mit SARS-CoV-2 infiziert.
Am 7. Januar 2020 gab der die Virusidentifizierung leitende chinesische Virologe Xu Jianguo (徐建国) bekannt, der Krankheitserreger sei ein bisher unbekanntes Coronavirus. Dies hätten Untersuchungen von Blutproben und Rachenabstrichen von 15 Erkrankten ergeben. Die WHO bestätigte diese Erkenntnis am 9. Januar 2020. Am 13. Januar 2020 wurde die komplette RNA-Genomsequenz eines Isolats des neuen Coronavirus in der NCBI-GenBank hinterlegt (GenBank-Nummer MN908947). Nahezu gleichzeitig wurde als erstes Nachweisverfahren für SARS-CoV-2 eine spezielle Polymerase-Kettenreaktion (PCR) publiziert – PCR ist eines der wichtigsten und zuverlässigsten Verfahren, um eine Infektion mit Viren nachzuweisen –, die Christian Drosten, Marion Koopmans, Chantal Reusken und ihre Virologie-Teams an der Charité Berlin, an der Erasmus-Universität Rotterdam und am niederländischen RIVM entwickelt hatten.
Eine phylogenetische Analyse der Genomsequenzen aus Umweltproben des Marktes (etwa von Oberflächen) zeigte, dass sie mit den Viren der ersten Patienten aus Wuhan sehr nahe verwandt sind. Nach einer Studie des Wuhan Hospitals hatte der erste identifizierte Patient den Markt nicht besucht. Keines der untersuchten Tiere vom Markt wurde positiv auf SARS-CoV-2 getestet, was die Annahme stützt, das Virus sei nicht dort auf den Menschen übergesprungen. Offenbar hatte sich das Virus zuvor unbemerkt unter Menschen etabliert. Der Markt könnte daher Schauplatz eines frühen Superspreader-Ereignisses gewesen sein.
Aus Modellierungen auf Basis der Untersuchung der Veränderungen des Erbmaterials RNA des Virus wird das erste Auftreten des Virus als wahrscheinlich zwischen Oktober und Anfang Dezember 2019 eingegrenzt. Das Verbreitungsmuster der verschiedenen unterscheidbaren Virusmutationen spricht für eine massenhafte weltweite Ausbreitung des Virus durch eine Vielzahl von verschiedenen Ausbreitungsereignissen.
Hongkongs größte englischsprachige Tageszeitung berichtete im März 2020 mit Verweis auf unveröffentlichte Regierungsdaten, als Patient null könnte sich ein 55-jähriger Mann aus der Provinz Hubei am 17. November 2019 infiziert haben. In einer im August 2021 veröffentlichten Studie konnte mit einem umfangreichen Genomvergleich eine vermutliche RNA-Sequenz der Ausgangsform („Stammvater“, en. progenitor: proCoV2) ermittelt werden, die (wie zu erwarten) vom Genom der real existierenden Referenzform etwas abweicht. Aus den Daten lässt sich ableiten, dass dieses Virus bereits einige Wochen vor den im Dezember 2019 entdeckten Erkrankungen Menschen infiziert hat. Nach Gesprӓchen mit chinesischen Medizinern schӓtzte die WHO die Anzahl der Corona-Patienten vor Dezember 2019 auf rund 1000 Personen. Zudem wurden 13 Virus-Stӓmme isoliert, die sich nicht alle dem Ausbruch in Wuhan zuordnen lassen. Es könnte sich bei 72.000 früheren Erkrankungen vom Oktober bis Dezember 2019 mit Symptomen wie Lungenentzündung, Grippe oder Fieber möglicherweise um COVID-19 gehandelt haben. Die nachträglich auf das Virus untersuchten 92 Proben fielen jedoch alle negativ aus.
Es sei nicht so einfach, gab die WHO im Frühjahr 2021 bekannt. Man gehe auch diesen, schon Wochen vor den ersten in Wuhan bekannt gewordenen Fällen nach, um zu ermitteln, ob man frühere Virenausbreitungen übersehen habe, doch die Testverfahren seien nicht standardisiert, nicht bestätigt, und es gebe weitere Herausforderungen. Im August 2021 erklärte die WHO, dass ihre neu gegründete Scientific Advisory Group for the Origins of Novel Pathogens (SAGO) diesen frühen Proben nachgehen werde und legte Wert darauf, sich nicht in Schuldzuweisungen zu üben.
Mit Stand September 2021 untersucht die WHO weiterhin die Herkunft des Virus, Ende November tagte die SAGO-Gruppe das erste Mal.
Bezeichnung
Das Virus SARS-CoV-2 wird im allgemeinen Sprachgebrauch (nach der Virusfamilie) als „neuartiges Coronavirus“, „neues Coronavirus“, „Coronavirus“ (zu Deutsch: Kranz- bzw. Kronenvirus) oder (in deutschsprachigen Ländern) nur als „Corona“ bezeichnet. Die von der WHO vom 13. Januar bis zum 11. Februar 2020 verwendete Bezeichnung „2019-nCoV“ galt nach deren Aussage nur vorläufig. Das National Center for Biotechnology Information (NCBI) nahm es als Wuhan seafood market pneumonia virus isolate Wuhan-Hu-1 in die Taxonomie-Datenbank auf. Das NCBI ist jedoch für Virusnamen und -klassifikationen nicht maßgebend. Das Virus wurde dort – ebenfalls vorläufig – als Wuhan seafood market pneumonia virus geführt; als Synonyme galten 2019-nCoV und Wuhan coronavirus.
Die WHO griff diverse Namensvorschläge nicht auf, die gemeinsam hatten, das Virus nach dem Ort seiner Erstidentifikation als Wuhan respiratory syndrome coronavirus (WRS-CoV) zu benennen. In der Vergangenheit hatte es Beschwerden gegeben, als Viren ihren Namen nach Ländern oder Regionen erhielten. (Beispiele: Marburg-Virus, MERS-CoV). Daher hatte die WHO 2015 Benennungen nach dem Entdeckungsort für unerwünscht erklärt. In der NCBI-Taxonomie-Datenbank aufgeführte Synonyme waren im Februar 2020: 2019-nCoV, COVID-19, COVID-19 virus, Wuhan coronavirus und Wuhan seafood market pneumonia virus.
Am 11. Februar 2020 gab die WHO bekannt, die durch das Virus verursachte Erkrankung als COVID-19 (oder „Covid-19“, für coronavirus disease 2019) benannt zu haben. Am selben Tag schlug die Coronavirus Study Group (CSG) des International Committee on Taxonomy of Viruses (ICTV) auf dem Preprint-Server bioRxiv für das Virus die Bezeichnung SARS-CoV-2 vor (für severe acute respiratory syndrome coronavirus 2). Dem widersprach eine Woche später eine Gruppe chinesischer Virologen, die stattdessen „HCoV-19“ („Humanes Coronavirus 2019“) einführen wollten. Damit würde der Virusname an den von der WHO bestimmten Namen der Krankheit COVID-19 angeglichen. Außerdem bestünde die Gefahr, das Virus SARS-CoV-2 mit dem Virus SARS-CoV zu verwechseln. Sie betonten, dass sich „2019-nCoV“ von dem SARS-Virus in biologischer und epidemiologischer Hinsicht unterscheide, ebenso wie die klinischen Symptome von COVID-19 und SARS verschieden seien. Letztlich wurde SARS-CoV-2 als offizieller Name veröffentlicht. Zur Unterscheidung wird der Erreger von SARS auch als SARS-CoV-1 bezeichnet.
Vergleichbare Diskussionen gibt es in Bezug auf die Bezeichnung der SARS-CoV-2-Varianten.
Merkmale

Systematik
Das Virus SARS-CoV-2 wurde mit seiner offiziellen Benennung und Klassifizierung durch das ICTV der Spezies Severe acute respiratory syndrome-related coronavirus zugeordnet. Diese Spezies enthält hunderte von Virusstämmen und Isolaten; die beiden Viren SARS-CoV (das seit 2020 auch SARS-CoV-1 genannt wird) und SARS-CoV-2 sind aber die einzigen in ihrer Spezies, die bisher als gefährliche Krankheitserreger (Virulenz) für den Menschen in Erscheinung traten. Unterhalb der Spezies gibt es keine offiziellen taxonomischen Einheiten (Taxa), sodass SARS-CoV-1, SARS-CoV-2 und andere Vertreter derselben Spezies nicht innerhalb von Unterarten gruppiert werden können. Benennungen unterhalb der Spezies sind von der Klassifizierung unabhängig, das heißt, dass gleiche Namensteile für Virusstämme und Isolate, wie z. B. „SARSr-CoV“, nicht in jedem Fall eine unmittelbare verwandtschaftliche Gruppierung ausdrücken.
In Gegensatz zu einer Einteilung von Viren unterhalb einer Art wird die Zuordnung von Virusarten bzw. der Spezies zu ihren übergeordneten Taxa durch das ICTV vorgenommen; die Spezies („Species“) Severe acute respiratory syndrome-related coronavirus gehört zur Untergattung („Subgenus“) Sarbecovirus und ist die einzige Spezies innerhalb dieser Untergattung. Die Untergattung Sarbecovirus gehört in die Gattung („Genus“) Betacoronavirus.
Innerhalb dieser Gattung gibt es weitere Untergattungen („Subgenera“), z. B. die Untergattung Merbecovirus, welche die Spezies Middle East respiratory syndrome-related coronavirus enthält. Innerhalb dieser Virusspezies ist in der Vergangenheit ein Virus, MERS-CoV, als krankmachender Erreger in Erscheinung getreten.
Die Beta-Coronaviren, wie die Mitglieder der Gattung Betacoronavirus auch gelegentlich genannt werden, enthalten letztlich bisher drei für den Menschen bekanntermaßen gefährliche Viren:
- SARS-CoV-1, das zur SARS-Pandemie 2002/2003 führte,
- MERS-CoV, das 2012 einen Ausbruch der entsprechenden Krankheit (MERS) bewirkte
- SARS-CoV-2, das Ende 2019 zu einem Ausbruch von COVID-19 führte, der wiederum der Ausgangspunkt einer COVID-19-Pandemie ist.
Die Gattung („Genus“) Betacoronavirus befindet sich in der Unterfamilie Orthocoronavirinae („Subfamily“) und diese wiederum in der Familie Coronaviridae („Family“). Der Trivialname für diese Familie, „Coronavirus“ in der Einzahl-Form und „Coronaviren“ in der Mehrzahl, bietet Verwechslungsmöglichkeiten, z. B. weil es bis 2009 eine offizielle Gattung gleichen Namens (Coronavirus) gab. Auf der Höhe dieses taxonomischen Ranges, Familie („Family“), ist die Zuständigkeit einer Arbeitsgruppe angesiedelt, der „Coronaviridae Study Group of the International Committee on Taxonomy of Viruses“, die auch die Benennung und Klassifizierung von SARS-CoV-2 vorgenommen hat.
Die Coronaviridae gehören zu den Cornidovirineae („Suborder“, Unterordnung) und diese zu den Nidovirales („Order“, Ordnung). Letztere wird noch in den virologischen Realm der RNA-Viren bzw. Riboviren (Riboviria) klassifiziert, da ihr Erbmaterial aus RNA besteht. Dadurch werden aber keine weiteren Verwandtschaftsbeziehungen im phylogenetischen Sinne ausgedrückt.
Es gibt also in der Systematik hinsichtlich der Kategorien eine Lücke zwischen dem niedrigsten offiziellen taxonomischen Rang, der Spezies („Species“, Virusart), und den Viren innerhalb der Spezies, welche die Krankheit COVID-19 als Erreger auslösen können, die aber keine benannten Ränge haben. Als Ersatz wurden anfangs die Begriffe „Klade“ und „Schwesterklade“ zur Adressierung von SARS-CoV-2 innerhalb seiner taxonomischen Spezies vorgeschlagen. Eine Klade berücksichtigt aber lediglich angenommene Verwandtschaftsbeziehungen, nicht aber die medizinischen Aspekte für die Mitglieder einer Gruppe von Viren; unter anderem deshalb wurde die sogenannte Pango-Nomenklatur entwickelt, bei der Abstammungslinien von SARS-CoV-2 nach epidemiologischen Kriterien bezeichnet und klassifiziert werden.
Aufbau
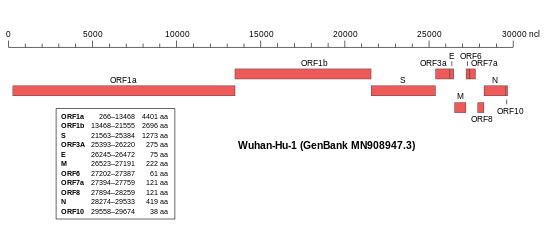


Das Virusgenom besteht, wie in Coronaviren üblich, aus einzelsträngiger RNA (ssRNA) mit positiver Polarität. Das Isolat Wuhan-Hu-1 (NCBI-GenBank-Nummer MN908947) umfasst 29.903 nt (Nukleotide) mit zwei 265 nt bzw. 229 nt langen untranslatierten Bereichen am 5′-Ende bzw. am 3′-Ende. Die putativen (vermuteten) Gene könnten für zehn Proteine codieren: ein 7096 Aminosäuren (AS) langes ORF1ab-Polyprotein (Replikase-Komplex), ein 1273 AS langes S-Glykoprotein – auch als Spike-Protein bezeichnet – ein 75 AS langes Hüllprotein (E für engl. envelope, vergleiche Virushülle), ein 222 AS langes Membran-Glykoprotein (M), ein 419 AS langes Nukleokapsid-Phosphoprotein (N) und weitere fünf Proteine (ORF3a, ORF6, ORF7a, ORF8 und ORF10). Die Abfolge der Gene entspricht jener des SARS-Virus und der aller anderen Coronaviren. Im November 2020 wurde nachträglich die Identifizierung eines „versteckten“ (überlappenden) Gens ORF3d bekannt gegeben.
Mit Stand 16. Februar 2020 gab es mehr als 40 vollständige Genomanalysen von SARS-CoV-2-Isolaten. Die Genomgröße liegt zwischen 29.825 und 29.903 nt. Der GC-Gehalt (der Anteil der Nukleinbasen Guanin und Cytosin) liegt bei 38,0 Mol-Prozent. Die beiden Virusisolate HKU-SZ-002a (NCBI-GenBank-Nummer MN938384) und HKU-SZ-005b (NCBI-GenBank-Nummer MN975262) stammen von Patienten einer Familie aus Shenzhen und unterscheiden sich lediglich durch zwei Nukleotide. Die Genomanalyse dieser beiden Isolate ergab, dass sie nahe verwandt mit den bei Fledermäusen (englisch bat) auftretenden SARS-CoV-ähnlichen Coronaviren bat-SL-CoVZXC21 (NCBI-GenBank-Nummer MG772934) und bat-SL-CoVZC45 (NCBI-GenBank-Nummer MG772933) sind, zu letzterem besteht eine Übereinstimmung in der Nukleotidabfolge von 89 %. Das Genom der beiden Fledermaus-Coronaviren wurde 2018 sequenziert, bat-SL-CoVZC45 wurde bei der Chinesischen Hufeisennase (Rhinolophus sinicus) aus der Familie der Hufeisennasen (Rhinolophidae) gefunden, die Wirtstiere wurden in Zhoushan in der ostchinesischen Provinz Zhejiang in den Jahren 2015 und 2017 untersucht.
Ein weiteres Virusisolat (WIV04, NCBI-GenBank-Nummer MN996528) von SARS-CoV-2 aus der bronchoalveolären Spülflüssigkeit eines der ersten Patienten zeigt ebenfalls phylogenetisch größte Ähnlichkeit mit einem bei einer anderen Fledermausart (Java-Hufeisennase, wissenschaftlich Rhinolophus affinis, englisch intermediate horseshoe bat, verbreitet in Indonesien (Java), Indien, Vietnam, China) in der chinesischen Provinz Yunnan isolierten Coronavirus BatCoV RaTG13; die Genomsequenzen stimmen zu 96,2 % überein. Auch eine am 27. Januar 2020 publizierte genetische Analyse verwies auf Fledermäuse als mutmaßlicher Ursprungswirt des Virus. Am 29. Januar 2020 wurde in der Fachzeitschrift The Lancet eine genetische Analyse von zehn Virusproben publiziert, die bei neun Erkrankten gewonnen worden waren. Demnach war die Genomsequenz aller zehn Proben zu 99,98 Prozent identisch, was darauf hinweist, dass die neu entdeckte Coronavirusvariante erst vor Kurzem auf den Menschen übergegangen ist. Die Genomsequenz stimmt zu 88 bzw. 87 % Prozent mit den Genomsequenzen der bei Fledermäusen auftretenden bat-SL-CoVZC45 und bat-SL-CoVZXC21 überein. Die zehn Proben zeigen hingegen nur rund 79 Prozent Übereinstimmung in der Genomsequenz zu SARS-CoV und rund 50 Prozent zu MERS-CoV. Die Ergebnisse der phylogenetischen Untersuchungen werden auch als phylogenetischer Baum, der die Verwandtschaftsverhältnisse von SARS-CoV-2 innerhalb der Coronaviridae zeigt, veranschaulicht. Eine darauf basierende Darstellung ist im Artikel Betacoronavirus zu finden.
Der Aufbau des Genoms sowohl der SARS-CoV-2-Isolate wie auch der genannten Fledermaus-Coronaviren ist typisch für Viren der Lineage B (Untergattung Sarbecovirus, englisch SARS-like Betacoronavirus) der Gattung Betacoronavirus. Aufgrund der genetischen Distanzen zu SARS-CoV und zu MERS-CoV wurde SARS-CoV-2 zunächst als eine in Bezug auf den Menschen neue, ihn infizierende Betacoronavirus-Spezies angesehen. Aufgrund der großen genetischen Übereinstimmung mit dem ursprünglichen SARS-Coronavirus hatte am 11. Februar 2020 die Coronavirus Study Group des ICTV jedoch vorgeschlagen, das neue Virus derselben Spezies Severe acute respiratory syndrome-related coronavirus zuzuordnen wie das bisherige.
Das Spike-Protein ist für die Bindung an die Wirtszelle verantwortlich. Funktionell wird es in zwei Untereinheiten, die S1-Domäne und die S2-Domäne unterschieden, die durch proteolytische Spaltung aus einem linearen Vorläufer entstehen. Die S1-Domäne vermittelt die Bindung an den Oberflächenrezeptor der Wirtszelle, die S2-Domäne vermittelt die Fusion der Zellmembran; durch Endozytose erfolgt dann der Eintritt des Virus in die Zelle. Das S-Gen von SARS-CoV-2 zeigt mit 75 % eine eher geringe Übereinstimmung in der Nukleotidsequenz mit den beiden Fledermausisolaten bat-SL-CoVZC45 und bat-SL-CoVZXC21 im Vergleich zur Genomanalyse. Insbesondere die Nukleotidsequenz, die für die S1-Domäne codiert, unterscheidet sich von diesen deutlich (68 % Übereinstimmung) und weist aber eine größere Ähnlichkeit mit der entsprechenden Nukleotidsequenz von BatCoV RaTG13 auf. Es wurde aufgezeigt, dass SARS-CoV-2 und SARS-CoV den gleichen Zellrezeptor nutzen, das Angiotensin-konvertierende Enzym 2 (ACE2). Dies konnte experimentell sicher nachgewiesen werden. (vgl. Krankheitsentstehung bei COVID-19)
Beim Vergleich des Genoms von SARS-CoV-2 mit dem verwandter Fledermaus-Coronaviren zeigten sich neben der bekannten Änderung am Spike-Protein zwei weitere „stille Mutationen“ (in den Nichtstrukturproteinen NSP4 und NSP16, siehe Coronaviridae §Genom), die zwar nichts an den kodierten Proteinen, jedoch die 3D-Faltung der RNA ändern. Dies könnte dazu beitragen, dass Infizierte (anfangs) zwar ansteckend, aber (noch) symptomfrei sind.
Morphologie
Coronaviren sind membranumhüllte RNA-Viren. In einer Zellkultur über mehrere Tage vermehrte Viren können nach Abtrennung durch Ultrazentrifugation für die Untersuchung im Transmissionselektronenmikroskop (TEM) vorbereitet werden; dabei wird eine Negativkontrastierung verwendet. Das TEM-Bild zeigt Virionen von kugelförmiger bis pleomorpher Gestalt mit einem Durchmesser von 60 bis 140 Nanometer (nm); nach anderer Quelle 91 ± 11 nm.
Auf der Oberfläche sind 9 bis 12 nm lange Spikes zu erkennen. Die Morphologie entspricht der anderer bekannter Vertreter der Familie der Coronaviridae. Die Wirtszellen, die im lichtmikroskopischen Bild einen cytopathischen Effekt aufweisen, können nach Fixierung und anschließendem Ultradünnschnitt (Dicke von 80 nm) ebenfalls mit dem TEM untersucht werden. Hier zeigen sich neben Virionen auch Einschlusskörperchen, die mit Viren gefüllte membrangebundene Vesikel im Cytoplasma enthalten.
Replikationszyklus

Der Replikationszyklus der Viren verläuft über neun Schritte (siehe Abbildung):
- Zunächst bleiben SARS-CoV-2-Virionen an speziellen Rezeptoren haften, die sich an der Oberfläche möglicher Wirtszellen befinden. Das geschieht, indem sich die Spike-Proteine der Vironen an die ACE2-Rezeptoren der Zellmembran binden. Der ACE2-Rezeptor der Wirtszellen ist deshalb ein möglicher Therapieansatz, um den Ausbruch einer COVID-19-Erkrankung nach einer Infektion mit dem Coronavirus zu verhindern. Ob weitere Moleküle der Zelloberfläche das Spike-Protein binden, ist noch nicht geklärt. Im Vergleich zu SARS-CoV hat das Spike-Protein eine RGD-Peptidsequenz entwickelt, womit Rezeptoren der Integrinfamilie ebenfalls als mögliche Bindungspartner in Frage kommen.
- Nach Bindung an den ACE2-Rezeptor spaltet die membranständige Serinprotease TMPRSS2 das virale Glykoprotein S, wodurch das Spike-Protein als fusogenes Protein aktiviert wird und der Eintritt in die Wirtszelle erfolgt. Auch TMPRSS2 ist ein potentieller Ansatzpunkt für ein wirksames Medikament.
- Die Erreger werden in die Wirtszelle aufgenommen (vereinfachte Darstellung).
- Vor Beginn der Virusvermehrung muss die Erbsubstanz (RNA) des Virus aus dem Kapsid freigesetzt werden (nur ein möglicher Weg dargestellt).
- Nun kann der eigentliche Vermehrungsvorgang erfolgen, die Replikation. Da SARS-CoV-2 über RNA positiver Polarität verfügt, kann die RNA direkt als „Bauanleitung“ für virusspezifische Proteine dienen, ähnlich zelleigener mRNA bei der Translation. Für die Wirtszelle ist die Virus-RNA praktisch nicht von eigener mRNA zu unterscheiden und der Proteinsyntheseapparat (Ribosomen) der Wirtszelle produziert so anhand der viralen RNA-Vorlage die virusspezifischen Proteine (S, M, E, N, RNA-Polymerase).
- Die RNA trägt die genetische Information des Virus. Sie wird als dessen Erbsubstanz in der Wirtszelle durch Kopieren vervielfältigt (RNA-Replikation). Dazu sind die Enzyme der Wirtszelle nicht in der Lage; diese Aufgabe übernimmt die virale RNA-Polymerase und stellt zahlreiche Kopien der gesamten Virus-RNA her.
- Sind in der Wirtszelle virale RNA-Kopien und Virusproteine in hinreichender Menge hergestellt, werden sie in das endoplasmatische Retikulum (ER) aufgenommen und lagern sich dort zu neuen Viren zusammen (Selfassembly).
- Die fertigen Viruspartikel werden als Golgi-Vesikel aus dem ER abgeschnürt (Knospung).
- Durch Exozytose gelangen die Viren aus der Wirtszelle und liegen nun als Virion vor, womit wiederum mögliche Wirtszellen infiziert werden können (siehe 1).
Umweltfaktoren
Ein weiterer Beschleunigungsfaktor für die Ausbreitung des Virus könnte in der Außentemperatur liegen, da sich das Virus laut einer chinesischen Studie bei 4 Grad Celsius als besonders persistent (langfristig aktiv) erwiesen hat. In der Luft liegt die kürzeste Überlebensdauer des Virus bei Raumtemperatur und mittlerer Feuchte, was mit den virentötenden Sauerstoffradikalen (ROS) zu tun haben könnte. Untersuchungen mit simuliertem Sonnenlicht vergleichbar mit der Sonneneinstrahlung an einem Sommertag ergaben eine Inaktivierung von rund 90 % der als Aerosol vorliegenden Viren binnen acht Minuten. Bei Innenraumbedingungen dauerte es mehrere Stunden, bis eine Inaktivierung dieser Größenordnung erreicht wurde. Eine Saisonalität von SARS-CoV-2 könnte in mittleren Breiten mit Temperaturen knapp über dem Gefrierpunkt von Wasser und einer Luftfeuchtigkeit von 40 bis 60 Prozent oder von 68 bis 88 Prozent einhergehen (also hierin zwei notwendige Bedingungen haben), während Bevölkerungsdichte, menschliches Verhalten, bevorzugte Aufenthaltsorte über Tag, medizinische Versorgung, Immunabwehr, Virusmutationen und Impfungen als maßgebliche Faktoren zu gelten haben, wenn es um den Verlauf und die Phasen dieser wie anderer Pandemien geht. In den Regionen der subtropischen Klimazone scheinen hohe Temperaturen die Ansteckung mit SARS-CoV-2 zu fördern; so etwa in Indien in der Zeit vor und während des Monsuns, wenn die Menschen wegen Hitze und Nässe zu Hause bleiben. Eine Übertragung durch Schmierinfektion wurde hingegen nicht beobachtet, ist aber nicht ganz auszuschließen. Im März 2021 publizierte die Wissenschaftsfachzeitschrift PNAS das Forschungsergebnis einer internationalen Forschergruppe unter der Leitung von Forschern der Technischen Universität München, laut dem eine hohe Pollenkonzentration in der Luft signifikant korreliert mit deutlich steigenden SARS-CoV-2-Infektionsraten.
Eine Auswertung von Daten aus 2.669 Kreisen in den Vereinigten Staaten ergab einen Saisonalitätseffekt, der mit der Luftfeuchtigkeit, kühleren Temperaturen und weniger UV-Einstrahlung korreliert. Es ließ sich eine jahreszeitlich bedingte Steigerung der effektiven Basisreproduktionszahl um rund 20 % nachweisen. Eine Untersuchung von 2021 beschäftigt sich mit der möglichen Infektion durch die Verwendung von Bargeld. Die Forscher kommen zum Ergebnis, dass die Infektionswahrscheinlichkeit als sehr gering eingeschätzt werden kann.
Virusvarianten
Entstehung
Seitdem das Coronavirus SARS-CoV-2 den menschlichen Organismus infiziert und sich explosionsartig in der Welt ausgebreitet hat, erwerben die „neuartigen“ Coronaviren trotz Korrekturaktivität der viralen Exonuklease eine zunehmende Anzahl von polymorphen Nukleotidsequenzen in verschiedenen Leserastern des viralen Genoms, anhand derer diese Varianten in sog. »Linien« (englisch lineages) unterteilt werden. Bei den Mutationen des Virus werden unterschieden:
- Synonyme Mutationen (eine Form stiller Mutationen), die sich nicht auf die codierten Proteine auswirken, da das veränderte Codon für dieselbe Aminosäure steht.
- Nichtsynonyme Mutationen mit Auswirkungen auf den Phänotyp (das Erscheinungsbild des Virus in all seinen Ausprägungen). Wichtig für die Entwicklung von Antikörpern und Impfstoffen ist es, herauszufinden, welche Teile der kodierten Proteine stabil bleiben und konserviert werden, damit die Medikamente nicht durch Mutation der Viren schnell wirkungslos werden.
Mutationen können die Infektiosität und Kontagiosität von SARS-CoV-2 verändern.
Wildtyp-Varianten
Die zu Beginn in China aufgetretenen Varianten werden als der Wildtyp bezeichnet; dazu gehört unter anderem das Isolat Wuhan Hu-1 (aus Variante B), das als Grundlage für die Entwicklung der mRNA-Impfstoffe von Biontech und Moderna verwendet wurde. Das komplette Genom von Wuhan Hu-1 wurde bereits im Januar 2020 frei veröffentlicht.

Mutationen und Varianten
Ira Deveson organisierte eine Untersuchung des ganzen Virus-Genoms von 157 mit SARS-CoV-2 infizierten Patienten. Die Geräte von Oxford Nanopore Technologies (ONT) ermöglichten sehr schnelles Sequenzieren. Ein überraschendes Ergebnis war, wie sehr die Virenproben variierten.
Mutation D614G ab Variante B.1
Im Frühjahr 2020 veränderte erstmals eine Mutation an der Position 614 des Genoms den Wildtyp-B von SARS-CoV-2. Die so entstandene Virusvariante-B.1, die auch als „Klade B.1" oder „Linie B.1" bezeichnet wird, wurde dominant, verbreitete sich seit März 2020 zunächst in Europa und dann weltweit. Dort ist im von der RNA codierten Spike-Protein die Asparaginsäure (D) durch Glycin (G) ersetzt. Die Mutation selbst wird daher als D614G bezeichnet und die Variante, die sich vom Wildtyp im Wesentlichen nur durch diese Mutation unterscheidet, in der Pango-Nomenklatur als B.1-Linie bezeichnet. Die Mutation D614G verursacht keine schwerere Erkrankung, erzeugt jedoch mehr Viruskopien und ist darum infektiöser und kontagiöser.
Fast alle heutigen für COVID-19-Erkrankung relevanten Varianten basieren auf der B.1-Linie und tragen die Mutation D614G (s. Abb., lila): So hat in der Neutralisationstiter-Untersuchung des Impfstoffherstellers Moderna vom Juni 2021 die deutliche Mehrheit der aufgelisteten Varianten (Alpha, Beta, Gamma, Delta, Epsilon, Eta, Iota und Kappa) diese Veränderung; nur die von der WHO nicht extra bezeichneten Varianten A.23.1-v1 und A.23.1-v2 (aus Uganda) sowie A.VOI.V2 (aus Angola) haben diese Veränderung nicht.
Zeitliches Verhalten und Ausbreitung
„Klassisch“ mit linearer Skala – absolute Anteile gut ablesbar, exponentielle Änderungen kaum.
(Beide Diagramme stellen exakt die gleichen Daten dar, mit unterschiedlichem Fokus.)
Logarithmische Skala – exponentielle Änderungen der Varianten gut erkennbar, die Anteile kaum.
(Beide Diagramme stellen exakt die gleichen Daten dar, mit unterschiedlichem Fokus.)
Einen Stammbaum der bis Ende Februar 2020 bekannten SARS-CoV-2-Isolate, der ihre Verwandtschaft untereinander zeigt, findet man bei Li et al. Die Isolate gliederten sich in zwei Hauptgruppen (L-Typ nach der Aminosäure Leucin und S-Typ nach Serin), was Anlass zur Vermutung gab, das Virus könnte sich in zwei (unterschiedlich infektiöse) Zweige aufgeteilt haben. Allerdings war es nach Meinung anderer Experten Anfang März 2020 noch zu früh, darüber eindeutige Aussagen machen zu können. Die in beiden Hauptzweigen des Stammbaums basal liegenden Isolate stammen aus Wuhan, was ein Beleg dafür war, dass das Virus dort seinen Ausgang nahm. Gleichwohl ist nicht ausgeschlossen, dass es einen unbekannten Vorläufer von anderswo, etwa aus der chinesischen Provinz Yunnan, in Tieren oder Menschen, gegeben haben könnte; auch das Einschleppen nach China durch den Import von Wirtstieren ist nicht auszuschließen (→ Herkunft und Wirtsspektrum).
Eine weitere Studie Anfang April 2020 machte drei Stämme A, B und C aus. Stamm A war dem Fledermausvirus BatCoV/RaTG13 am ähnlichsten und scheint sich von Wuhan aus weltweit verbreitet zu haben; in Festlandchina selbst war aber Stamm B vorherrschend, der außer in China auch andernorts in Ostasien verbreitet war. Stamm C war der hauptsächliche Typ in Europa.
Das Virus schien zu Beginn relativ langsam zu mutieren – ein bis zwei Mutationen pro Monat wurden beobachtet (zum Vergleich: Influenzaviren mutieren zwei- bis viermal so häufig). Das bedeutete zum einen, dass es per Genomanalyse keine sehr hohe Auflösung bezüglich der Ausbreitungswege des Virus gab, zum anderen ließ es darauf hoffen, dass eine nach überstandener Krankheit erworbene Immunität lange (monatelang) anhält. Allerdings hatten isländische Virologen von deCODE Genetics (isländisch Íslensk erfðagreining) bis zum 24. März 2020 vierzig verschiedene Mutationen allein bei Infizierten aus diesem Land identifiziert. Eine der Betroffenen war mit zwei verschiedenen Ausprägungen von SARS-CoV-2 coinfiziert. Die im Westen dominierende Form des Virus, die sich ab Februar 2020 in Europa stark ausbreitete und von dort auch in andere Länder, hat eine Mutation D614G im Spike-Protein und weicht damit von der Wuhan-Variante ab. Insbesondere hat diese Mutation vier- bis fünfmal so viele Spikes auf der Oberfläche des Virus.
In einer italienischen Studie vom Juli 2020 wurden zu diesem Zeitpunkt sechs SARS-CoV-2-Varianten unterschieden. Stamm G ist in Europa am häufigsten, dieser ist seit Ende Februar 2020 weiter mutiert in die Stämme GR und GH. Der ursprüngliche Stamm L aus Wuhan wird immer weniger gefunden, wie auch der Stamm V. Ein Stamm S wurde in einigen Gebieten der USA und Spaniens gefunden.
Der Anteil der SARS-CoV-2-Varianten Alpha (B.1.1.7), Beta (B.1.351) und Gamma (P.1) an den Infektionszahlen kann anhand einer neuen Überprüfungsmethode, die von einem gemeinsamen Nukleotid der drei Varianten ausgeht, schneller ermittelt werden. Diese Untersuchungen sollen zweimal im Februar und einmal Anfang März 2021 in Deutschland wiederholt werden.
Nomenklatursysteme der Varianten

Das SARS-CoV-2 Virus besteht aus ca. 30.000 Nukleotiden. Durch Mutationen gibt es eine riesige Anzahl von Varianten, von denen nur ein Bruchteil relevant ist, und sie nimmt immer weiter zu. Es gibt mehrere Bezeichnungssysteme, mit denen die vielzähligen Varianten geordnet werden. Zumeist wird die Einteilung nach der Abstammung (Entwicklungslinien, Kladen) vorgenommen.
Das bekannteste System ist die Pango-Nomenklatur, in dieser stammt z. B. die Variante B.1.1.7 von B.1.1 ab, die wiederum vom B.1 und schließlich von B abstammt. Die Pango-Nomenklatur bezeichnet die frühesten Entwicklungslinien als Variante A (mit Isolat Wuhan/WH04/2020) und B (mit Wuhan-Hu-1), die beide zu Beginn in China auftraten. Obwohl die Variante B etwas früher isoliert und nachgewiesen wurde, vermutet man, dass die mit A bezeichnete Variante die ursprünglichere ist. Auch in einer Variante, wie z. B. der Variante B oder Untervariante B.1, haben die einzelnen Viren nicht exakt dasselbe Genom. Erst wenn es hinreichend bedeutende Veränderungen gibt und diese auch in der Natur auftreten, wird in der dynamischen Pango-Nomenklatur eine neue (Unter-)Variante dafür definiert. Zu einer Variante gibt es also etliche etwas unterschiedliche Isolate. In der Variante B ist z. B. das Isolat Wuhan Hu-1 enthalten, das am 26. Dezember 2019 in Wuhan beprobt („sampled “) wurde.
Daneben gibt es auch die Kladeneinteilung nach Nextstrain und die nach GISAID sowie Bezeichnungen nach den Mutationen im Spike-Protein, z. B. D614G oder 501Y.V1. Die Einteilung nach Abstammung hat das Problem der konvergenten Mutationen, d. h. eine Mutation, die ursprünglich zur Unterscheidung zwischen zwei Abzweigungen diente, kann in der Linie, die sie ursprünglich nicht hatte, später dennoch eintreten.
Klassifizierung und Bezeichnung gemäß WHO
Um eine Stigmatisierung von Ländern zu vermeiden, bezeichnet die Weltgesundheitsorganisation (WHO) die gemäß eigener Einstufung besorgniserregenden oder beobachtungsbedürftigen Varianten des Coronavirus SARS-CoV-2 seit dem 31. Mai 2021 mit Buchstaben aus dem griechischen Alphabet. Dabei wurden die Buchstaben Ny und Xi übergangen, da Ny (englisch nu) mit dem englischen Wort new verwechselt werden könne und Xi ein gängiger Familienname sei, sodass die nach My als nächstes benannte Variante die Bezeichnung Omikron (englisch Omicron) erhielt.
Nach diesem Schema heißt die zuerst in Großbritannien nachgewiesene Virusvariante B.1.1.7 nun Alpha und die in Südafrika entdeckte B.1.351 nun Beta. Die Untervarianten B.1.617.1 und B.1.617.2 der erstmals in Indien nachgewiesenen Virusvariante B.1.617 werden von der WHO mit Kappa und Delta bezeichnet. Die vormals „brasilianische Variante“ genannte Virusvariante P.1 (B.1.1.28.1) erhielt die Bezeichnung Gamma. Die bereits eingeführten wissenschaftlichen Nomenklaturen für Virusvarianten behalten laut WHO ihre Berechtigung.
Die Weltgesundheitsorganisation und die CDC verwenden folgende Kategorien, denen die SARS-CoV-2-Varianten zugeordnet werden:
Variant of High Consequence (VOHC)
Variante von hoher Bedeutung, diese Kategorie wird von der US-amerikanischen CDC verwendet. Dieser Klasse werden Varianten zugeordnet bei:
- Nachweis von Fehldiagnosen,
- signifikanter Verringerung der Wirksamkeit des Impfstoffs,
- einer unverhältnismäßig hohen Anzahl von Krankheitsausbrüchen trotz Impfung,
- einem sehr geringen Schutz gegen schwere Krankheitsverläufe trotz Impfung,
- eine deutlich verringerte Wirksamkeit der Pharmazeutika mit Notfallzulassung oder regulärer Zulassung,
- schwerwiegenden klinischen Erkrankungen und Anstieg der Krankenhausaufenthalte.
Variant of Concern (VOC)
Besorgniserregende Variante, wird von der Weltgesundheitsorganisation (WHO) eingestuft. Eine Variant of Concern (VOC) von SARS-CoV-2 entspricht der Definition einer VOI (Variant of Interest), zudem wurde durch eine vergleichende Bewertung nachgewiesen, dass eine oder mehrere der folgenden Veränderungen von globaler Bedeutung für die öffentliche Gesundheit zutreffen:
- Zunahme der Übertragbarkeit oder Verschlechterung der COVID-19-Epidemiologie (Verbreitung in der Bevölkerung); ODER
- Zunahme der Virulenz (Ansteckung, Vermehrung, Krankheitsschwere) oder Veränderung des klinischen Krankheitsbildes (Symptome); ODER
- Nachlassende Wirksamkeit der öffentlichen Gesundheits- und Sozialmaßnahmen oder der verfügbaren Diagnostika (Nachweismethoden), Impfstoffe, Therapeutika (Arzneimittel).
Die Varianten Alpha B.1.1.7, Beta B.1.351 waren ab Dezember 2020, Gamma P.1 ab Januar 2021 als Variant of Concern klassifiziert. Im Mai 2021 kam Delta B.1.617.2 hinzu, im November 2021 Omicron B.1.1.529.
Omikron: B.1.1.529

Die Omikron-Variante ist eine Untervariante der Variante B.1.1. Sie wurde am 9. November 2021 erstmals identifiziert und Ende November mit B.1.1.529 bezeichnet. Die WHO stufte sie am 24. November 2021 als Variant Under Monitoring ein, zwei Tage später wurde sie mit dem griechischen Buchstaben Omikron als Variant of Concern bezeichnet.
Diese Variante zeichnet sich durch rund 30 Aminosäureänderungen im Spikeprotein aus. Davon befinden sich 15 im Teil des Proteins, das an den Rezeptor auf der menschlichen Zelle bindet. Darunter sind auch Mutationen, die bei anderen Virussubtypen mit einer Erhöhung der Übertragbarkeit und einem Unterlaufen der Immunantwort in Verbindung gebracht werden.
Bei der Variante Omikron wurde eine höhere Wachstumsrate als bei der Delta-Variante bestätigt. Bei Personen, die weder geimpft sind noch vorher mit einer anderen Variante infiziert waren, ist das Hospitalisierungs-Risiko nur etwa ein Drittel so hoch wie bei der Variante Delta. Die Impfung reduziert weiterhin die Hospitalisierungen. Die Variante Omikron führt laut RKI auch bei vollständig Geimpften und Genesenen häufig zu Infektionen, die weitergegeben werden können. 25 Wochen nach Grundimmunisierung mit zwei Dosen lag die Impfstoffeffektivität gegen Hospitalisierung nur noch bei 44 % (95-%-KI 30–54), zwei Wochen nach Auffrischungsimpfung bei 92 %, zehn Wochen und später bei 83 % (95-%-KI 78–87).
Die Variante steht in Zusammenhang mit einem Anstieg der COVID-19-Fälle in der südafrikanischen Provinz Gauteng im November 2021. Reiseassoziierte Fälle wurden im selben Monat 2021 in Hongkong, Belgien, Israel und Deutschland nachgewiesen. Bis 9. Dezember wurden weltweit über 2000 Fälle in 60 Staaten gemeldet, im Januar 2022 dominierte Omikron mit etwa 90 % weltweit.
Ehemals zirkulierende VOC
In der Unterkategorie „Previously circulating VOCs“ fasst die WHO ehemals zirkulierende besorgniserregende Varianten zusammen:
Alpha: B.1.1.7
Im Dezember 2020 wurde in der britischen Grafschaft Kent die Variante Alpha (B.1.1.7, auch mit VOC-202012/01, VUI-202012/01 und N501Y.V1 bezeichnet) des Coronavirus SARS-CoV-2 mit den Mutationen 69-70del, P681H und N501Y (letztere am Spike-Glykoprotein) festgestellt. Diese hat nach Mitteilung der britischen Regierung vom 19. Dezember 2020 gegenüber den anderen Varianten die Oberhand gewonnen. Die New and Emerging Respiratory Virus Threats Advisory Group (NERVTAG) ist der Ansicht, dass sich der neue Virusstamm, zu dem auch Varianten mit der Mutation P681H oder der Deletion von H69 und V70 im Spike-Protein gehören, schneller verbreiten kann; die molekulare Ursache könnte im Fall der N501Y-Mutation die bessere Bindung an den menschlichen Zellrezeptor ACE2 des viralen Spike-Proteins sein, während die Deletion von H69 und V70 die Bindung mancher menschlicher Antikörper an das Spike-Protein verschlechtern könnte. Mit Stand März 2021 wurde die Variante Alpha in 82 Ländern nachgewiesen. Von Ende Januar 2021 bis zur zweiten Märzwoche stieg der Anteil der Variante Alpha an den in Deutschland positiven SARS-CoV-2-Proben von 6 % auf 72 %. Eine Auswertung britischer Daten zeigt eine Zunahme des Reproduktionsfaktors R um 43–90 % im Vergleich zum Wildtyp. Ähnliche Beobachtungen liegen aus den USA und Dänemark vor. Eine Kohortenstudie aus dem Vereinigten Königreich kam auf Basis von rund 100.000 Krankheitsverläufen zu dem Schluss, dass die Variante das Sterberisiko um rund 64 % gegenüber dem Wildtypvirus erhöhe. Am 9. März 2022 stufte die WHO diese Variante als „Previously circulating VOC“ zurück.
Beta: B.1.351
Am 18. Dezember 2020 meldete das südafrikanische Gesundheitsministerium die Entdeckung der Variante Beta (B.1.351, auch N501Y.V2). Sie weist ebenfalls die N501Y-Mutation auf, deren Auftreten anscheinend unabhängig vom Auftreten in der südenglischen Grafschaft Kent ist. Diese Variante soll möglicherweise noch ansteckender sein und auch bei jungen Leuten einen schweren Krankheitsverlauf verursachen können. Am 8. Februar 2021 wurde diese Variante in mehr als 30 Ländern nachgewiesen.
In Österreich gab es im Februar 2021 Hinweise darauf, dass sich in Teilen des Bundeslands Tirol die Variante Beta verstärkt ausbreitete. Zu der Zeit wurde etwa die Hälfte der dort durch eine Mutation verursachten Infektionen auf diese Variante des Virus zurückgeführt. 80 % der Neuinfektionen mit SARS-CoV-2 würden vom ursprünglichen Virus, dem Wildtyp, verursacht und jeweils 10 % von der Variante Alpha (B.1.1.7) oder Variante Beta (B.1.351), wie die Virologin Dorothee von Laer von der Medizinischen Universität Innsbruck der Nachrichtenagentur dpa mitteilte. Am 9. März 2022 stufte die WHO diese Variante als „Previously circulating VOC“ zurück.
Gamma: P.1
Die Variante Gamma, in Pango-Nomenklatur P.1 alias B.1.1.28.1 bezeichnet, wurde erstmals im November 2020 in Brasilien nachgewiesen. Am 10. Januar 2021 wurde gemeldet, dass sie im brasilianischen Bundesstaat Amazonas zirkuliert. Sie ähnelt den Varianten Alpha und Beta und weist ebenfalls die N501Y-Mutation auf. Die Untervariante Gamma stammt von der Variante B.1.1.28 ab und wird auch als 501Y.V.3 bezeichnet. Am 22. Januar 2021 wurde bekannt, dass die Variante Gamma erstmals in Deutschland gefunden worden war. Bei einem aus Brasilien kommenden Hessen, der am Flughafen Frankfurt eingereist war, konnte eine Infektion mit der Variante mittels PCR-Test nachgewiesen werden. Eine DNA-Sequenzierung stand zu diesem Zeitpunkt jedoch noch aus. Ebenso wie die Varianten Alpha und Beta steht diese Variante im Verdacht, ansteckender zu sein als der Wildtyp des Coronavirus SARS-CoV-2. Laut Aussage der Virologin Sandra Ciesek von Mitte Januar 2021 gebe es keine Hinweise darauf, dass diese Varianten schwerere Verläufe verursachen als der Wildtyp des Virus.
Im Département Moselle, das an das Saarland, Rheinland-Pfalz und Luxemburg grenzt, waren auffallend hohe Häufungen der Varianten Beta und Gamma registriert worden. Der französische Gesundheitsminister Olivier Véran sagte, vom 8. bis 11. Februar 2021 seien 300 Fälle dieser Varianten nachgewiesen und in den Tagen zuvor weitere 200 Fälle. Am 9. März 2022 stufte die WHO diese Variante als „Previously circulating VOC“ zurück.
Delta: B.1.617.2

Die Delta-Variante B.1.617.2 des Coronavirus SARS-CoV-2 wird mit ihren Untervarianten AY.* seit dem 11. Mai 2021 von der WHO zu den „besorgniserregenden Varianten“ (englisch Variants of concern, VOC) gezählt. Ende August 2021 betrug ihr Anteil in Deutschland 99,3 % aller sequenzierten Proben.
Mit der Delta-Variante Infizierte stecken im Mittel mehr als doppelt so viele andere Menschen an als beim Ursprungs-Virus. Die Dauer von der Ansteckung bis zum Nachweis der Viren ist dabei im Schnitt von sechs auf vier Tage verkürzt, die Virusmenge etwa 1200-mal höher. Nach Risikoeinschätzung der englischen Gesundheitsbehörde Public Health England (PHE) Anfang Juni 2021 kann die Delta-Variante häufiger zu schwereren COVID-19-Erkrankungen führen als die Alpha-Variante (B.1.1.7) des Virus. Mit der Variante Delta Infizierte haben ein etwa doppelt so hohes Risiko wie bei Alpha, wegen COVID-19 in ein Krankenhaus eingewiesen zu werden.
Impfungen verhindern Infektionen mit der Delta-Variante mindestens in etwa der Hälfte der Fälle. Das Risiko, schwer krank zu werden oder zu sterben, ist im Mittel für ungeimpfte Personen mehr als zehnmal höher als bei geimpften, im höheren Alter lässt die Schutzwirkung der Impfungen nach. Mit den bisherigen COVID-19-Impfstoffen übertragen geimpfte Personen das Virus in ähnlichem Maße wie ungeimpfte, so die WHO im August 2021. Am 7. Juni 2022 stufte die WHO diese Variante als „Previously circulating VOC“ zurück.
Variant of Interest (VOI)
Variante von Interesse, wird von der Weltgesundheitsorganisation (WHO) eingestuft als Variant of Interest (VOI):
- Genetische Veränderungen (Mutationen), von denen vorausgesagt wird oder bekannt ist, dass sie Viruseigenschaften wie Übertragbarkeit, Krankheitsschwere, Immunescape (Ausweichen vor dem Immunsystem), diagnostische oder therapeutische Möglichkeiten beeinflussen; UND
- Verursachung einer signifikanten Übertragung in der Bevölkerung oder mehrerer COVID-19-Ausbrüche, in mehreren Staaten mit steigender relativer Prävalenz (Häufigkeit) bei steigender Fallzahl im Zeitverlauf oder anderen offensichtlichen epidemiologischen Auswirkungen (Verbreitung in der Bevölkerung), die ein aufkommendes Risiko für die globale öffentliche Gesundheit erwarten lassen.
Ehemals zirkulierende VOI
In der Unterkategorie „Previously circulating VOIs“ fasst die WHO ehemals zirkulierende Varianten von Interesse zusammen:
Epsilon: B.1.427/B.1.429
Am 19. Januar 2021 wurde die Variante B.1.427/B.1.429 (Epsilon) mit Mutation L452R in Kalifornien bekannt, die sich von der Variante Alpha (B.1.1.7) unterscheidet. Sie wurde erstmals im März 2020 nachgewiesen, war im März 2021 als „Variant of Interest“ klassifiziert, ab Juli zurückgestuft zur Beobachtung in „Alerts for Further Monitoring“ und im November 2021 als „Formerly monitored variant“ aus der Kategorisierung genommen.
Eta: B.1.525

Die Variante B.1.525 (Eta) wurde im Dezember 2020 in mehreren Staaten erstmals nachgewiesen und vereint Genveränderungen der Varianten Alpha (B.1.1.7) und Beta (B.1.351). Am 24. Dezember 2020 wurde diese neue Variante von SARS-CoV-2 in Nigeria entdeckt, die sich von den Varianten Alpha und Beta unterscheidet. Sie ist in mehreren Ländern nachgewiesen worden, unter anderem in Dänemark, Italien, Nigeria, Norwegen, Kanada, Großbritannien und den USA. Am 9. März 2021 wurde gemeldet, sie sei in Deutschland am Flughafen BER erstmals nachgewiesen worden. Das Unternehmen Centogene, das die Probe analysiert hat, teilte kurz darauf mit, die Variante sei auch schon in anderen Proben nachgewiesen worden. Die Variante Eta enthält die Mutation E484K, die WHO stufte sie Mitte März 2021 als „Variant of Interest“ (VOI) ein. Die charakteristischen Mutationen des Spike-Proteins sind Q52R, A67V, 69/70del, 144del, E484K, D614G, Q677H und F888L. Die WHO stufte diese Variante am 20. September 2021 zurück auf Variant Under Monitoring und nahm sie Ende Dezember 2021 als „Formerly monitored variant“ aus der Kategorisierung.
Iota: B.1.526

Die Variante B.1.526 (Iota) wurde erstmals im November 2020 in den USA nachgewiesen und erhielt Ende März 2021 ihre Bezeichnung. Seit Februar 2021 grassierte in New York City diese neue Variante, die Gemeinsamkeiten mit den Varianten Beta (B.1.351) und Gamma (P.1) hat. Am 10. März 2021 wurde bekannt, dass fast 40 % der in örtlichen Laboren untersuchten COVID-Infektionen auf die Variante Iota zurückzuführen sind. Die charakteristischen Mutationen des Spike-Proteins sind L5F, T95I, D253G, D614G und A701V, ggfs. E484K oder S477N. Die WHO stufte diese Variante am 20. September 2021 zurück auf Variant Under Monitoring und nahm sie Ende Dezember 2021 als „Formerly monitored variant“ aus der Kategorisierung.
Kappa: B.1.617.1
Die Variante B.1.617.1 (Kappa) ist eine Untervariante von B.1.617 (Indien). Ihr erstes Auftreten wurde auf Oktober 2020 zurückverfolgt, Anfang April 2021 erhielt sie ihre offizielle Bezeichnung. Die WHO stufte diese Variante am 20. September zurück auf Variant Under Monitoring und nahm sie Ende Dezember 2021 als „Formerly monitored variant“ aus der Kategorisierung.
Lambda: C.37


Die Lambda-Variante C.37 alias B.1.1.1.37 wurde Mitte Juni 2021 von der Weltgesundheitsorganisation (WHO) als „Variant of Interest“ (VOI) eingestuft und Lambda genannt. Sie breitet sich seit August 2020 in Südamerika aus; als Ursprungsland gilt Peru. Auf der Preprint-Plattform bioRxiv wurden im Juli 2021 zwei (wissenschaftlich noch nicht begutachtete/gegengeprüfte) Studienergebnisse zu Lambda veröffentlicht, die zu unterschiedlichen Schlüssen kommen. Die erste von Mikrobiologen der New York University vorgestellte Studie beschreibt Lambda als infektiologisch unspektakulär und machtlos gegen die durch COVID-19-Impfstoffe erzeugten Antikörper. Die zweite von einem Wissenschaftler der Universität Tokio vorgestellte Studie stuft Lambda dagegen durch die „einzigartige 7-Aminosäure-Deletionsmutation“ RSYLTPGD246-253N als resistent gegen die (bis dahin) gängigen COVID-19-Impfstoffe ein und schreibt der Lambda-Variante eine höhere Infektiosität als dem Urtyp von SARS-CoV-2 zu, wegen der Mutationen T76I und L452Q. Die charakteristischen Mutationen des Spike-Proteins sind G75V, T76I, del247/253, L452Q, F490S, D614G und T859N. Am 9. März 2022 stufte die WHO diese Variante als „Previously circulating VOI“ zurück.
My: B.1.621


Die Variante My B.1.621 (englisch Mu) wurde zuerst im Januar 2021 in Kolumbien nachgewiesen, beinhaltet die Untervariante B.1.621.1 und machte dort Ende August bereits 39 Prozent der Infektionsfälle aus. Bis Juli 2021 hatte die britische Gesundheitsbehörde Public Health England (PHE) 16 Fälle mit der vergleichsweise neuen Variante bestätigt und am 21. Juli 2021 zur „Variant under Investigation“ (VUI) erklärt, es bestand noch kein Verdacht auf eine unkontrollierte kollektive Verbreitung. Offenbar stehe ein Großteil der neuen Fälle in Verbindung mit Überseereisen.
Die WHO bezeichnete sie mit dem zwölften Buchstaben des griechischen Alphabets „Mu“ (englisch) bzw. „My“ (deutsch) und stufte sie Ende August 2021 als „Variant of Interest“ (VOI) ein, nachdem sie in 39 Staaten gefunden wurde, wenn auch nur mit einem Anteil von 0,1 % an den Varianten weltweit, Mitte September 2021 in Europa nur sehr vereinzelt. Sie teilte mit, dass diese Variante Mutationen mit möglichen Resistenzen gegen Corona-Impfstoffe aufweise, ähnlich wie die Beta-Variante oder noch stärker, auch als alle anderen VOC- und VOI-Varianten, so eine vorveröffentlichte Studie vom September 2021. Charakteristische relevante Mutationen des Spike-Proteins sind E484K, N501Y, D614G und P681H, die teilweise auch in den VOC-Varianten Alpha, Beta und Gamma zu finden sind, zudem die Mutationen T95I, Y144T, Y145S und das eingefügte 146N in der N-terminalen Domäne. Am 9. März 2022 stufte die WHO diese Variante als „Previously circulating VOI“ zurück.
Variant under Investigation (VUI)
Variante unter Beobachtung ist eine Art nationale Unterkategorie der Variant of Interest (VOI) für neu entdeckte Varianten. Sie wird von nationalen Gesundheitssystemen wie Public Health England (PHE) genutzt, um frühzeitig – noch vor Kategorisierung durch die WHO – Viren mit möglicherweise besorgniserregendem Potenzial für die genauere Nachverfolgung zu kategorisieren.
Variant Under Monitoring (VUM)
Variante unter Überwachung, wird von der Weltgesundheitsorganisation (WHO) eingestuft als Variant Under Monitoring (VUM) (bis Sommer 2021 als Alerts for Further Monitoring benannt, bei gleicher Definition). Bei diesen Varianten gibt es genetische Veränderungen (Mutationen) mit Hinweisen darauf, dass eine Beeinflussung der Viruseigenschaften vorliegen könnte, die möglicherweise künftig ein erhöhtes Risiko erwarten lassen, wobei noch keine klaren Beweise für phänotypische oder epidemiologische Auswirkungen vorliegen, sodass die Variante eine stärkere Überwachung erfordert, bis eine Neubewertung erfolgen kann, sobald neue Beweise vorliegen.
Varianten dieser Kategorie werden zurzeit nicht als weltweites Risiko angesehen und werden zum Teil durch nationale Gesundheitsbehörden weiter als Variant of local interest eingestuft, sie werden auch von der WHO weiter überwacht. Sollte sich ihre Charakteristik ändern, würden sie neu bewertet und eingestuft.
Unklassifizierte Varianten
- Im Oktober 2020 wurde die Variante B.1.617 im indischen Bundesstaat Maharashtra erstmals nachgewiesen. Bis Ende April 2021 wurde sie in weiteren Staaten nachgewiesen, darunter dem Vereinigten Königreich, Deutschland, der Schweiz, Belgien, den Vereinigten Staaten, Australien und Singapur. In Deutschland machte sie Anfang Mai 2021 rund 2 % der sequenzierten Proben aus. Bis Ende Juni stieg der Anteil auf 37 % der sequenzierten Proben. Bei B.1.617 wurden durch Mutation drei Aminosäuren im Spike-Protein ausgetauscht. Die Mutationen E484K und E484Q führten zu einer reduzierten Wirksamkeit der humoralen Immunantwort, während die Mutation L452R sowohl eine reduzierte Wirksamkeit der humoralen als auch der zellulären Immunantwort zufolge hatte. Die Klade B.1.617 wird in die Subkladen B.1.617.1 (Kappa-Variante) und B.1.617.2 (Delta-Variante) unterteilt, wobei letztere nicht die Mutation E484Q aufweist.

- Die Variante C.1.2 alias B.1.1.1.1.2 ist eine Untervariante von C.1. Sie wurde Mitte Mai 2021 erstmals in den Provinzen Mpumalanga und Gauteng in Südafrika identifiziert, Ende Juli offiziell benannt und breitet sich nach Vermutungen von Forschern ähnlich schnell aus wie die Delta-Variante. Im August 2021 verbreitete sie sich in sechs von neun Regionen Südafrikas, zudem in der Demokratischen Republik Kongo, Mauritius, Neuseeland und Botswana. In Europa wurde C.1.2 in Portugal und der Schweiz erstmals nachgewiesen. Im März 2022 stufte die WHO diese Variante als „Formerly monitored variant“ zurück.

- Die Variante B.1.640 ist eine Untervariante der Variante B.1. Sie wurde am 28. September 2021 erstmals identifiziert, am 10. November benannt und am 22. November als Variant Under Monitoring eingestuft. Ihre zugehörige Untervariante B.1.640.2 wurde am 8. Dezember benannt. Französische Forscher um Didier Raoult vom Institut IHU Méditerranée Infection wiesen sie bei zwölf Patienten im Südosten Frankreichs nach. Der vermutlich in Frankreich zuerst Infizierte kam von einer Reise aus Kamerun zurück. Das Team um Raoult schrieb, es sei „zu früh, um über virologische, epidemiologische oder klinische Eigenschaften der neuen Variante zu spekulieren“. Die Variante B.1.640.2 wird in den Medien nach dem Méditerranée Infection University Hospital Institute (IHU) in Marseille inoffiziell auch als „IHU-Variante“ bezeichnet. Ende Mai 2022 stufte die WHO diese Varianten als „Formerly monitored variant“ zurück.
Herkunft und Wirtsspektrum
Seit dem Bekanntwerden der Viruskrankheit werden verschiedene Tiergruppen als Ursprung oder zumindest Überträger des Erregers diskutiert. Eine molekulare Datierungsschätzung mittels Genom-Vergleich der verschiedenen SARS-CoV-2-Isolate legt einen Ursprung der Virusvariante im November 2019 nahe. Van Dorp und ihre Kollegen ermittelten aufgrund phylogenetischer Analysen der verschiedenen Virusvarianten Anfang Mai 2020, dass das Virus zwischen dem 6. Oktober und dem 11. Dezember 2019 auf den Menschen übergesprungen sein dürfte.
Eine vergleichende Studie zum Infektionsrisiko von SARS-CoV-2 / COVID-19 wurde im August 2020 von Joana Damas et al. vorgelegt. Danach ist das Bindungspotential des Spike-Proteins an den jeweiligen ACE2-Rezeptor bei Primaten (Mensch, Bonobo, Gemeiner Schimpanse, Westlicher Flachlandgorilla) am größten, bei folgenden Spezies dagegen sehr gering: Kalifornischer Seelöwe, Hausmaus, Amerikanerkrähe und Mississippi-Alligator. Insgesamt können mehr als 60 Säugetier-Arten von SARS-CoV-2 infiziert werden, darunter auch Füchse, Yaks, Riesenpandas und Koalas. (Stand: 6. November 2020).
Siehe auch: Institut für Virologie Wuhan
Fledertiere und Schuppentiere

Hufeisennasen – möglicherweise mehrere höhlenbewohnende Arten – waren das Reservoir des Erregers SARS-CoV-1, der die SARS-Pandemie 2002/2003 ausgelöst hatte, mit dem Larvenroller (Paguma larvata, englisch masked palm civet) als möglichem Zwischenwirt zwischen Fledertier und Mensch. Seitdem wurden verschiedene weitere Betacoronaviren (insbesondere auch SARS-artige der Untergattung Sarbecovirus) vor allem bei Fledertieren, aber auch bei Menschen gefunden.
BatCoV RaTG13
Anfang 2020 wurde für ein Virus, welches BatCoV RaTG13 genannt wurde, die große Übereinstimmung seiner Genomsequenz zu derjenigen von SARS-CoV-2 mit einem Wert von 96,2 % angegeben. Verglichen mit anderen zu SARS-CoV verwandten Coronaviren („SARSr-CoVs“), die zu dieser Zeit durch eine Arbeitsgruppe um die Virologin Shi Zhengli in die Untersuchung einbezogen werden konnten, zeigte RaTG13 die höchste Übereinstimmung mit dem Virus SARS-CoV-2 (welches damals noch 2019-nCoV genannt wurde). Das Virus RaTG13 ist also mit SARS-CoV-2 eng verwandt und daher ein Anhaltspunkt, um die Abstammung von SARS-CoV-2 einzugrenzen.
Die Entdeckungsgeschichte vom Virus RaTG13, welches im Jahr 2013 aus Kot der Fledermaus-Art Rhinolophus affinis isoliert wurde, ist indirekt an das Auftreten von schweren Atemwegserkrankungen beim Menschen im Jahr 2012 gekoppelt, wie erst Ende 2020 in einem Nachtrag (Addendum) mitgeteilt wurde. Die Vorfälle in 2012 haben das Interesse an der Erforschung der Viren bei Fledermäusen in einer Kupfermine in Tongguan gefördert. Allerdings wird in den entsprechenden Publikationen an keiner Stelle ein Nachweis des Virus RaTG13 als Auslöser von Erkrankungen beim Menschen erwähnt und es werden dort auch keine Untersuchungen beschrieben, die einen solchen Nachweis erbracht haben könnten. Im Folgenden wird die Entdeckungs- und Publikationsgeschichte von RaTG13 dargestellt:
- Im Jahr 2012 wurden Reinigungsarbeiten zur Beseitigung von Fledermauskot in einer Kupfermine vorgenommen, die sich in oder bei der Stadt Tongguan (通关镇; Karte) im Landkreis Mojiang (墨江县) in der chinesischen Provinz Yunnan befindet (bzw. befand). Es ist in diesem Zusammenhang zu Lungenentzündungen gekommen, die zum Teil tödlich endeten. Die Erkrankten waren am 26. und am 27. April 2012 im nächstgelegenen Krankenhaus aufgenommen worden und es wurden dort im weiteren Verlauf Proben von den Patienten gesammelt. Diese Serumproben wurden auf das Vorhandensein verschiedener Viren getestet und es wurden keine dieser Viren gefunden. Die Proben wurden jedoch nicht auf RaTG13 und auch nicht auf SARS-CoV-2 getestet, da diese Viren 2012 noch nicht bekannt waren. In den Jahren 2012 und 2013 wurden Stuhlproben von Fledermäusen im stillgelegten Minenschacht im Landkreis Mojiang entnommen (erste Probenahme im August 2012), um sie hinsichtlich des Virenspektrums zu untersuchen; die Ergebnisse dieser Forschung sind Anfang 2016 veröffentlicht worden. Die Stuhlproben von sechs Fledermausarten sind nach Genabschnitten des RdRp-Gens durchsucht worden, die zu den Gattungen Alphacoronavirus und Betacoronavirus passen, sodass die dabei gefundenen (und zumeist nicht klassifizierten) Coronaviren am wahrscheinlichsten diesen beiden Gattungen zugeordnet werden könnten. Ein Betacoronavirus-Kandidat, der zudem als eng mit SARS-CoV verwandt eingestuft wurde, war das Virusisolat „RaBtCoV/4991“, dessen teilweise Sequenz des RdRp-Gens im Jahr 2013 in GenBank (Zugriffsnummer KP876546) hinterlegt wurde. Später, im Jahr 2018, konnte durch verbesserte Methoden die nahezu vollständige Genomsequenz ermittelt werden, diese wurde aber erst 2020 – im Zuge der entsprechenden Publikation – in GenBank (Zugriffsnummer MN996532) mit einer neuen Benennung („RaTG13“ statt „RaBtCoV/4991“) hinterlegt. Im Nachtrag (Addendum) zur eigentlichen Publikation wird erklärt, dass der Virusname die Art des Wirtes (also Rhinolophus affinis), den Fundort (also Tongguan) und das Jahr der Isolation (also 2013) wiedergeben sollte, weshalb für die entsprechende Publikation eine Umbenennung von der ursprünglichen Proben-Nr. des Virusisolates („4991“) zum Virusnamen „RaTG13“ vorgenommen wurde.
SARS-CoV-ähnliche Coronaviren
Bis 2017 wurden in den Höhlen in Yunnan SARS-CoV-ähnliche Coronaviren in folgenden Fledermausspezies gefunden: in Hufeisennasenarten bei der Java-Hufeisennase (Rhinolophus affinis, en. intermediate horseshoe bat), der Chinesischen Hufeisennase (R. sinicus) und der Großen Hufeisennase (R. ferrumequinum); sowie in der Stoliczka-Dreizackblattnase (Aselliscus stoliczkanus, en. Stoliczka's trident bat).
In Kot einer Horn-Hufeisennase (Rhinolophus cornutus, en. little Japanese horseshoe bat) aus der Präfektur Iwate im Norden der japanischen Hauptinsel Honshū vom Jahr 2013 wurde im Herbst 2020 ein Rc-o319 genannter Sarbecovirus-Stamm gefunden, dessen Genom zu 81 % mit dem von SARS-CoV-2 übereinstimmt.
BatCoV RaTG13 und SARS-CoV-2
Aufgrund der Ähnlichkeit der Bindungsstelle (en. receptor binding domain, RBD) des Spike-Proteins an den menschlichen Rezeptor ACE2 (hACE2) gilt inzwischen das Virus-Isolat BatCoV RaTG13 (gefunden in Java-Hufeisennasen Rhinolophus affinis, englisch intermediate horseshoe bat in Yunnan, in Bruchstücken auch bei erkrankten und verstorbenen Minenarbeitern aus Yunnan 2016), als wichtiger Kandidat für den Ursprung von SARS-CoV-2, auch wenn nicht klar ist, ob die Übertragung direkt erfolgte. Die Übereinstimmungen der Gesamtgenomsequenzidentitӓt zwischen RaTG13 und SARS-COV-2, festgestellt beim Screening durch eine veröffentlichte Pan-CoV-2-PCR-Methode beträgt 96 %.
Zu Beginn der Pandemie kannte man praktisch keine nah mit SARS-CoV-2 verwandte Viren. Die hochaffine Bindung des SARS-CoV-2 Spike-Proteins an menschliches ACE2 ist höchstwahrscheinlich das Ergebnis einer natürlichen Selektion an einer menschlichen oder menschenähnlichen ACE2, die eine optimale Bindungslösung gestattet. Dass die Genetik des Spike-Proteins von SARS-CoV-2 so gut zum Menschen passt, wird immer wieder als Argument für einen Labor-Ursprung des Virus benutzt.
Schuppentiere
Nachdem in Malaiischen Schuppentieren (Manis javanica, en. Sunda pangolin) Coronaviren mit hoher genetischer Übereinstimmung zum SARS-CoV-2 gefunden wurden (Manis-CoV, genauer Pan_SL-CoV_GD/P1L, Isolate SRR10168377 und SRR10168378), gerieten diese in Verdacht der Ursprung der Pandemie zu sein, obwohl Schuppentiere Einzeltiere sind, die relativ kleine Populationsgrößen aufweisen, aber trotz Verbots in China gehandelt werden (Rote Liste gefährdeter Arten). Die Übereinstimmung betrug in diesem Fall 90 % über das gesamte Genom, aber 99 % in einer spezifischen Region des Spike-Proteins (S-Protein), die es dem Virus erlaubt, an die ACE-Rezeptoren der menschlichen Zellen zu binden. Interessanterweise ist das in den Java-Hufeisennasen (R. affinis) isolierte Virus RaTG13 gerade in diesem Genom-Abschnitt zu SARS-CoV-2 mit nur 77 % Übereinstimmung vergleichsweise unterschiedlich. Dies bedeutet, dass die aus den Malaiischen Schuppentieren isolierten Coronaviren in menschliche Zellen eindringen können, das aus Java-Hufeisennasen isolierte jedoch nicht. Außerdem ist dieses Ergebnis verträglich mit der Annahme, dass SARS-CoV-2 das Ergebnis einer Rekombination der RNA-Moleküle zweier verschiedener Viren sein könnte, eines dem RaTG13 aus Fledermäusen von Yunnan, das andere dem Pan_SL-CoV_GD aus den Schuppentieren von Guangdong nahestehend. Dann wäre SARS-CoV-2 entstanden als eine neue Chimäre aus zwei Viren, die diesen beiden Linien jeweils sehr nahestanden. Diese Annahme wurde durch eine weitere Studie von Xiaojun Li und Kollegen (Duke University, Los Alamos National Laboratory, University of Texas, El Paso und New York University) Ende Mai 2020 unterstützt.
Zwar besitzen Coronaviren – anders als etwa Influenzaviren – ein unsegmentiertes Genom (monopartit), d. h. nur ein einziges Nukleinsäuremolekül (hier RNA). Eine Rekombination von Segmenten als Ganzes (Reassortment) ist also im Gegensatz zu diesen nicht möglich. Insbesondere um den Ursprung des alten SARS-Virus SARS-CoV-1 zu erklären, wurde bereits früher bei dieser Virusfamilie ein Rekombinationsmechanismus, und zwar innerhalb des (einzigen) Genom-Segments, beschrieben (homologe Rekombination). Eine solche Rekombination kann, egal ob segmentiertes oder unsegmentiertes Genom, zu einem neuen Virus führen, das eine neue Wirtsspezies befallen und krank machen kann. Das Rekombinationsereignis kann daher zum Ausgangspunkt einer neuen Epidemie werden, wie es bei SARS vermutet (und bei Influenza stets befürchtet) wird. Voraussetzung ist die Doppelinfektion (Koinfektion) eines (einzelnen) Wirtsindividuums durch die beiden Ausgangsviren. Allerdings bleibt bislang (Stand 2. Juni 2020) ungeklärt, in welcher Spezies die hypothetische Doppelinfektion stattgefunden haben könnte, und unter welchen Umständen dies geschehen sein könnte. Bei den konfiszierten Schuppentieren, die in Quarantӓnezentren untergebracht wurden, konnten hochspezifische SARS-CoV-2-Antigene festgestellt werden.
Alternatives Szenario
Als alternatives Szenario, das ohne Rekombination auskommt, wird verschiedentlich etwa folgendes vorgeschlagen: Die gemeinsamen Vorfahren von RaTG13 und SARS-CoV-2 stammen danach ursprünglich von den Schuppentier-Coronaviren ab, von deren SARS-CoV-2-ähnlichstem Stamm sie sich vor mehr als 140 Jahren trennten. Diese Linie spaltete sich vor etwa 40–70 Jahren erneut auf: eine Linie verblieb in Fledermäusen und verlor dort die Bindungsfähigkeit ihres Spike-Proteins an das menschliche ACE2 (hACE2). Die andere behielt diese Fähigkeit und sprang zuletzt als SARS-CoV-2 auf den Menschen über. Die verschiedenen Möglichkeiten werden auch von Halloy et al. in einem PrePrint vom Juli 2020 diskutiert. Auch Boni et al. vertreten Ende Juli 2020 die Ansicht, dass SARS-CoV-2 nicht direkt aus einer Rekombination von Fledermaus- und Schuppentier-Coronaviren hervorgegangen ist, sondern dass sich seine Entwicklungslinie von der des Fledermausvirus RaTG13 vor ca. 50 Jahren getrennt hat.
Weiteres zu Nilflughunden siehe unten (Abschnitt Weitere Wirbeltiere).
Anfang Dezember 2020 wurde erstmals über Funde SARS-CoV-2-ähnlicher Coronaviren bei Fledermäusen außerhalb Chinas berichtet. Neben dem oben erwähnten Fund von Rc-o319 bei der Horn-Hufeisennase aus Japan könnte man bei zwei im Jahr 2010 eingefrorenen Exemplaren der Kochang-Hufeisennase (Rhinolophus shameli, en. Shamel's horseshoe bat) aus dem Norden Kambodschas fündig geworden sein, die Genom-Analyse ist aber erst zu 70 % abgeschlossen (Stand 6. Dezember 2020). Die Ergebnisse von Fledermausstudien waren jedoch im Allgemeinen beruhigend. Eine Untersuchung der ACE2-Rezeptoren in den Zellen von 46 Fledermausarten ergab, dass die Mehrheit schlechte Wirte waren. Einige Arten, wie z. B. Fruchtfledermäuse (Rousettus aegyptiacus), die infiziert wurden, konnten die Infektion auf andere Fledermäuse übertragen.
Marderhunde als mögliche Zwischenwirte

Laut Christian Drosten könnten Marderhunde (Nyctereutes procyonoides, eine Fuchsart) möglicherweise die gesuchten Zwischenwirte sein. Auch das ursprüngliche SARS-Virus (SARS-CoV-1) wurde in Marderhunden gefunden, die wegen ihres Fells in China gezüchtet werden und somit als Überträger auf den Menschen in Frage kommen.
Haustiere als Wirte
Haushunde und Hauskatzen waren die ersten Tiere, bei denen es im Haushalt ihrer Besitzer zu Übertragungen von Mensch zu Tier kam. Testtiere beider Arten wurden daher auch in Laborexperimenten infiziert, um den Verlauf der Krankheit und mögliche Rückübertragungen auf den Menschen zu erforschen. Einer im September 2020 publizierten Studie zufolge gibt es keine Hinweise für eine Rückübertragung der Viren auf den Menschen, wohl aber Belege dafür, dass die Immunantwort von infizierten Tieren beider Arten diese vor einer zweiten Infektion schützt. Hunde und vor allem Katzen stecken sich offenbar relativ häufig bei ihren mit SARS-CoV-2 infizierten Besitzern an. Darauf weisen zwei Untersuchungen hin. So berichtet die kanadische Tiermedizinerin Dorothee Bienzle, dass sie bei 67 % der untersuchten Katzen und bei 43 % der Hunde Antikörper fand, was auf eine durchgemachte Infektion hinweist. Die Tiere hatten mit infizierten Menschen zusammengelebt. Laut WHO gab es bereits im März 2020 Hinweise, dass Haustiere SARS-CoV-2 nicht als Träger weiterverbreiten. Jedoch können einige andere Viren aus der Virusfamilie Coronaviridae auch bei Haustieren Erkrankungen auslösen, z. B. die beiden Alphacoronaviren CCoV (Hunde) und FCoV (Katzen).
Nachfolgend einige Beispiele für die Erkrankungen bei Haustieren.
Hunde
Am 28. Februar 2020 gab die Regierung Hongkongs bekannt, erstmals einen Hund positiv auf das Virus getestet zu haben, der im Haushalt seiner infizierten Halter lebte. Die WHO bestätigte, die SARS-CoV-2-Proben seien „schwach positiv“ getestet worden. Obwohl bei dem Hund das Virus im Blut nachgewiesen werden konnte, löste es bei ihm keine klinisch nachweisbaren Hinweise auf eine Erkrankung aus. Das Tier wurde zuletzt am 12. und 13. März 2020 mit negativem Befund auf SARS-CoV-2 getestet, so dass seine Quarantäne beendet und es dem Besitzer zurückgegeben wurde. Zwei Tage nach Ende der Quarantäne verstarb der Hund, ohne dass ein direkter Zusammenhang mit dem Virusbefall nachweisbar war.
Mitte März 2020 wurden in Hongkong zwei weitere Hunde positiv auf SARS-CoV-2 getestet, die ebenfalls ohne auffällige Symptome einer Infektion waren. Aus Japan wurde im September 2020 bekannt, dass dort zwischen April und August vier Hunde von an SARS-CoV-2 erkrankten Haltern positiv getestet, ohne auffällige Symptome einer Infektion isoliert und nach wiederholtem, negativem Test an ihre gesundeten Halter zurückgegeben worden waren.
Mitte April 2020 erschien ein Artikel über die Möglichkeit von streunenden Hunden als Zwischenwirt für die Übertragung von Sarbecoviren (RaTG13, Pangolin-CoV) von Wildtieren (Fledermäusen, Schuppentieren) auf den Menschen. Eine wichtige Rolle spielt dabei das Zinkfingerprotein ZAP.
Wenn Viren in einen Organismus eindringen, dann wehrt er sich. Diese „Kampfspuren“ kann man später am Virus nachweisen bzw. an der Art, wie es sich verändert. Dies wurde von Professor Xuhua Xia untersucht. Dabei stellte er fest, dass nur die Coronaviren von Hunden (CCoVs) die gleiche Reaktion bei den Viren verursacht hatten wie im Fall des neuen Sars-CoV-2 und beim ursprünglichen Fledermausvirus BatCoV RaTG13.
Bei einer experimentellen Studie wurden drei Hunde mit dem Virus infiziert. Keiner der Hunde zeigte klinische Zeichen einer Infektion und es konnte auch keine Ausscheidung von vermehrungsfähigem Virus nachgewiesen werden.
Katzen
In Lüttich (Belgien) wurde Ende März 2020 die Hauskatze eines Infizierten positiv auf SARS-CoV-2 getestet. Das Tier litt vorübergehend an Durchfall, Erbrechen und erschwerter Atmung. Eine Ende März 2020 in Hongkong bei einer Hauskatze nachgewiesene Infektion verlief hingegen symptomlos. Antikörpernachweise hatten zuvor in Wuhan ergeben, dass dort auch Katzen infiziert worden waren. Zudem wurde mehrfach in Laborexperimenten belegt, dass infizierte Katzen die Viren an andere Katzen weitergeben können. Es besteht der Verdacht, dass eine Katze das Virus zwischen Bewohnern eines Altenheims in Bayern übertragen haben könnte, obwohl sie voneinander isoliert waren. Eine weitere infizierte Katze wurde in Barcelona untersucht. Das Tier war wegen einer Herzerkrankung eingeschläfert worden, jedoch ergab die Autopsie, dass es nicht an, sondern mit SARS-CoV-2 am Herz erkrankt war. Auch in der Schweiz wurde das Virus Ende 2020 bei einer Katze nachgewiesen.
Eine experimentelle Studie, bei der sieben Katzen infiziert worden waren, zeigte, dass diese rund fünf Tage übertragbares Virus ausschieden und auch andere Katzen infizieren konnten. Keine der untersuchten Katzen zeigte klinische Zeichen einer Infektion. Die durchgemachte Infektion schützte die Tiere im Falle einer Reexposition mit dem Virus. Ein Team unter der Leitung des Virologen Bu Zhigao führte Proben des SARS-CoV-2-Virus in die Nase von fünf Hauskatzen ein. Als zwei der Tiere sechs Tage später eingeschläfert wurden, fanden die Forscher virale RNA sowie infektiöse Viruspartikel in ihren oberen Atemwegen. Die anderen drei infizierten Katzen wurden neben nicht infizierten Katzen in Käfige gesetzt. Das Team entdeckte später virale RNA in einer dieser exponierten Katzen, was darauf hindeutet, dass es das Virus aus Tröpfchen kontrahierte, die von den infizierten Katzen ausgeatmet wurden. Alle vier infizierten Katzen produzierten auch Antikörper gegen SARS-CoV-2. Die Überwachung auf SARS-CoV-2 bei Katzen sollte als Teil der Bemühungen zur Eliminierung von COVID-19 beim Menschen betrachtet werden. Als Sporadische Infektionsquelle bei Menschen könnten Katzen nicht ausgeschlossen werden, sagte Jan Felix Drexler, Virologe am Charité-Krankenhaus in Berlin. Laut chinesischen Berichten wurden seit 2019 ungefähr 30.000 Wild,- Zucht- und Haustiere wissenschaftlich auf entsprechende Infektionen getestet. Lediglich im März 2020 entdeckte man dabei in Wuhan einige wahrscheinlich infizierte Katzen.
Im November 2021 starben im Lincoln Children’s Zoo in Lincoln, Nebraska, drei Schneeleoparden an den Folgen einer Infektion mit SARS-CoV-2, die rund einen Monat zuvor bei den Tieren diagnostiziert worden war.
Marderverwandte
Im April und Mai 2020 wurden erstmals Infektionen und Erkrankungen von Amerikanischen Nerzen (Neovison vison, wie im Englischen auch Mink genannt) festgestellt, und zwar in mehreren niederländischen Nerz-Farmen. Die erkrankten Nerze zeigten ähnliche Symptome wie erkrankte Menschen: Atemwegsbeschwerden, Probleme mit dem Verdauungstrakt, erhöhte Sterblichkeit. Auch in der vom Feinstaub belasteten Luft der Tierhaltungen wurde virale RNA nachgewiesen. Das Virus sei – so die anfängliche Vermutung – von infizierten Mitarbeitern eingeschleppt und danach von Tier zu Tier weitergegeben worden. Detaillierte Analysen des genetischen Codes der in den Farmen und im Umland der Farmen umlaufenden SARS-CoV-2-Varianten erbrachten zudem Anhaltspunkte dafür, dass sich zwei infizierte Mitarbeiter der Farmen bei den Nerzen angesteckt haben und dass zudem mehrere im Bereich der Farmen frei laufende Katzen ebenfalls „farm-typische“ SARS-CoV-2-Varianten aufwiesen, weswegen auch sie als mögliche Überträger von Viren auf die Nerze infrage kommen. Auch gab es Hinweise darauf, dass das Virus zwischen Mensch und Amerikanischem Nerz hin und zurück gesprungen ist, dass also eine Übertragung zoonotisch (von Nerz auf Menschen) möglich ist; untersucht wurden Ausbrüche auf 16 Nerzfarmen. Nach Ansicht der WHO-Expertin Maria Van Kerkhove ist das Risiko einer Ansteckung des Menschen durch ein solches Tier jedoch nur „sehr begrenzt“. Eine ausführliche Stellungnahme mit Empfehlungen zum Umgang mit Nerzen hat die Europäische Gesundheitsbehörde (ECDC) am 12. November 2020 abgegeben.
Im US-Bundesstaat Utah wurden zwischen Juli und September 2020 – nach auffälligen Häufungen von Todesfällen in mehreren Zuchtbetrieben – ebenfalls infizierte Nerze entdeckt. Ein wilder Nerz, der in Utah positiv getestet wurde, „könnte nur die Spitze des Eisbergs sein“, sagte Sarah Hamer, Epidemiologin und Tierärztin an der Texas A & M University in der College Station. „Je mehr wir schauen, desto mehr könnten wir finden.“ Mitte Oktober 2020 wurde bekannt, dass Massenkeulungen vorgenommen wurden. So wurden allein im US-Bundesstaat Utah fast 10.000 Tiere getötet, in Spanien über 92.000 und (von denen 90 % mit SARS-CoV-2 infiziert gewesen sein sollen) in den Niederlanden über eine Million. Anfang November wurde von amtlichen Stellen in Dänemark angekündigt, sämtliche im Land gehaltenen – bis zu 17 Millionen Nerze – zu töten. Vorausgegangen waren Erkenntnisse über Mutationen des Virus in Nerzen, gegen die einige der in Entwicklung befindlichen Impfstoffe gegen das Virus beim Menschen voraussichtlich nicht wirksam wären. Die Nerzzucht in Dänemark ist ein bedeutender Wirtschaftszweig mit jährlich rund 17 Millionen Fellen in rund 1100 Zuchtfarmen, auf denen die Tiere auf engem Raum in Käfigen gehalten werden. Bei den Nerzen in Dänemark wurde eine Virusvariante („Cluster 5“) entdeckt. Mitte November teilte die dänische Regierung mit, dass diese Variante ausgemerzt sei. Insgesamt war es bis November 2020 in sechs Ländern zu Ausbrüchen in Nerzfarmen gekommen. Bis Januar 2021 wurde das Virus in Nerzfarmen von acht Ländern in der EU/im EWR nachgewiesen. Dem Friedrich-Loeffler-Institut (FLI) zufolge waren für Deutschland keine besonderen Schutzmaßnahmen nötig, da es wegen des Verbots der Haltung von Nerzen als Pelztiere in Deutschland keine Nerzfarmen gibt. In Wuhan wurden zehn Marktstände gefunden, an denen wilde oder gezüchtete Tiere aus Farmen in Südchina verkauft wurden, darunter Kaninchen, Zibetkatzen, Waschbären und Frettchen. Peter Daszak forderte eine Untersuchung der Bestände und Mitarbeiter dieser Farmen, ob sich dort möglicherweise noch Antikörper gegen das Virus finden lassen.
Durch Laborexperimente von Kim Young-Il et al. an der Universität Chungbuk in Südkorea wurde belegt, dass Frettchen empfänglich für eine SARS-CoV-2-Infektion sind und die Viren auch an Artgenossen weitergeben können. Das FLI bestätigte aufgrund eigener Tests den Befund der Universität Chungbuk und wies zugleich darauf hin, dass auch Nilflughunde empfänglich für eine SARS-CoV-2-Infektion sind, Schweine und Hühner hingegen nicht. Insbesondere die Empfänglichkeit von Frettchen sei ein wichtiger Befund, „da sie als Modelltiere für die Infektion des Menschen zur Erprobung von Impfstoffen oder Medikamenten eingesetzt werden könnten“. Eine vorveröffentlichte Studie exponierte Frettchen entweder über die Nase oder die Luftröhre mit SARS-CoV-2, wobei die Infektion bei jüngeren Frettchen nur über die Nase Fuß fassen konnte. Alle Frettchen zeigten keine beobachtbaren Krankheitszeichen, jedoch eine Hyperplasie der Lymphknoten der Bronchien.
Ein experimentelles Vakzin gegen COVID-19 wurde an den in ihrem Bestand stark gefährdeten (Endangered, IUCN 3.1) Schwarzfußiltissen erprobt.Finnland entwickelt einen Impfstoff für Marderhunde und Amerikanische Nerze, um in den Pelztierfarmen keine Massenkeulungen vornehmen zu müssen. Auch Russland entwickelt einen Impfstoff für Nerze, Katzen und Nagetiere.
Primaten
Ein Übergang von SARS-CoV-2 vom Menschen auf Menschenaffen wurde erstmals Anfang 2021 nachgewiesen, und zwar bei den Gorillas im San Diego Zoo Safari Park. Laut einer Mitteilung der Zooverwaltung hatten zwei Gorillas am 6. Januar 2021 zu husten begonnen, weswegen Fäkalien der Tiere auf das Virus getestet wurden. Aufgrund der positiven Befunde wurden weitere Tests durch das veterinärmedizinische Labor des U.S. Department of Agriculture durchgeführt, die ebenfalls positive Befunde erbrachten. Abgesehen vom Husten waren in der Gorilla-Gruppe keine gesundheitlichen Auffälligkeiten zu beobachten.
Weitere Befunde:
- Im Jahr 2016 wurde bei Schimpansen im Tai-Nationalpark (Elfenbeinküste) eine Infektion mit dem Humanen Coronavirus OC43 (HCoV-OC43, ein Betacoronavirus aus der Untergattung Embecovirus, Spezies Betacoronavirus 1) beobachtet, das bei Menschen milde erkältungsartige Symptome hervorruft. Diese zeigten auch die Schimpansen. Um SARS-CoV-2-Übertragungen zu vermeiden, wurde daher im Frühjahr 2020 (insbesondere für Wildhüter) empfohlen, zu den Schimpansen einen Mindestabstand von 7 bis 10 Meter zu halten und auch gegenüber den Tieren gegebenenfalls Quarantänezeiten einzuhalten.
- Eine chinesische Forschergruppe um Chuan Qin stellte im März 2020 Ergebnisse ihrer Untersuchungen an Rhesusaffen zur Verfügung. Hierbei ging es insbesondere um die Frage der Infektiosität nach überstandener Erkrankung. Auch eine im Mai 2020 in Science publizierte Studie an Rhesusaffen berichtete von „schützender Immunität“ nach erstmaliger Erkrankung.
- Niederländische Forscher berichteten im März 2020 in Science, dass SARS-CoV-2 bei Javaneraffen eine „COVID-19-ähnliche Krankheit“ verursache, weswegen diese Tiere als Modell für das Testen von vorbeugenden und therapeutischen Strategien geeignet seien.
Weitere Wirbeltiere
Im New Yorker Bronx Zoo wurde Anfang April 2020 ein erwachsener Tiger positiv auf SARS-CoV-2 getestet, nachdem bei ihm trockener Husten und keuchender Atem aufgefallen waren, jedoch keine Atemnot. Weiterhin wiesen auch zwei Löwen und fünf Tiger ähnliche Symptome auf, weswegen auch bei ihnen eine Infektion mit SARS-CoV-2 vermutet wurde. Infiziert wurden die Tiere vermutlich von einem asymptomatischen Bediensteten des Zoos. Wenige Tage nach dem Auftreten von Krankheitszeichen erholten sich die Tiere wieder. Im Joburg Zoo in Johannesburg (Südafrika) infizierte sich im Juli 2020 ein Puma bei einem infizierten Tierbetreuer. Chinesische Forscher berichteten im April 2020 in der Fachzeitschrift Science, dass sich das Virus in Hunden, Schweinen, Hühnern und Enten nur schlecht („poorly“) vermehre, und bestätigten, dass Frettchen und Katzen infiziert werden können. Auch Goldhamster, die nach einer Infektion mit SARS-CoV-1 nur sehr schwache Symptome entwickelt hatten und daher als Modelltiere ungeeignet waren, ließen sich im Labor mit SARS-CoV-2 infizieren, zeigten deutliche Symptome und wiesen hohe Viruskonzentrationen in Lunge und Darm auf.
Wie bereits im Abschnitt Marderverwandte erwähnt, hatte das Friedrich-Loeffler-Institut (FLI) aufgrund eigener Tests solche Befunde bestätigt: Nilflughunde sind neben Frettchen (im Gegensatz zu Schweinen und Hühnern) empfänglich für eine SARS-CoV-2-Infektion.
Diese Ergebnisse wurden durch eine Studie von Kore Schlottau (WHO) et al. (veröffentlicht im Juli 2020) ein weiteres Mal bestätigt und vertieft. Getestet wurden Nilflughunde (Rousettus aegyptiacus, englisch fruit bats), Frettchen (von den Autoren als Mustela putorius bezeichnet), Hausschweine (Sus scrofa domesticus) und Haushühner (Gallus gallus domesticus). Die Hausschweine und Haushühner erwiesen sich auch hier als nicht empfänglich für SARS-CoV-2. Als die Forscher begannen, Schweine und Ferkel künstlich mit SARS-CoV-2 zu infizieren, stellten sie fest, dass es sich nicht gut replizierte. Sieben von neun Nilflughunden erkrankten zunächst an Rhinitis, das Virus wanderte mit weiterem Fortschreiten der Erkrankung über die Luftröhre teilweise bis in die Lunge. Bei den Frettchen wurde zwar eine noch effizientere Virusreplikation, aber bis auf eine mögliche leichte Rhinitis keine Krankheitssymptome beobachtet. Sie entwickelten wie auch die Nilflughunde Antikörper gegen SARS-CoV-2.
Laut einer Studie des Friedrich-Loeffler-Instituts (FLI) zeigen Rinder eine geringe Empfänglichkeit gegenüber SARS-CoV-2. In den USA wurden bei Untersuchungen von wild lebenden Weißwedelhirschen Infektionen mit verschiedenen Abstammungslinien nachgewiesen. Die Studienautoren gehen von mehreren verschiedenen Eintragungsereignissen aus und schätzten Infektionen von Tier zu Tier als wahrscheinlich ein.
Während im Labor infizierte Mäuse offenbar keine Krankheitssymptome entwickeln, war es Y.-C. Wang und Kollegen in China möglich, bei C57BL / 6-Labormäusen mit CRISPR / Cas9 das ACE2 der Mäuse (mACE2, murines ACE2) durch das des Menschen (hACE2, humanes ACE2) zu ersetzen. Die hACE2-Mäuse zeigten Virusreplikation von SARS-CoV-2 in ihren Lungen, der Luftröhre und im Gehirn. Auch der Verdauungstrakt war betroffen, so wie es bei manchen menschlichen Patienten beobachtet wird. Die Mäuse scheinen damit geeignet, um etwa einen Impfstoff zu testen, bevor er Menschen verabreicht wird – eine Alternative zur Methode, die Wirkung eines Mittels auf künstlich mutierten Sarbecoviren zu testen, wie jüngst bei Remdesivir und SARS-CoV-RdRp / SARS-CoV-2-RdRp (altes SARS-Virus mit RdRP-Gen von SARS-CoV-2) geschehen. Durch die Varianten Beta und Gamma können Mäuse nachweislich infiziert werden.
Risikogruppe nach Biostoffverordnung
Für Beschäftigte, die durch ihre berufliche Tätigkeit mit Infektionserregern in Kontakt kommen können, gilt in Deutschland die Biostoffverordnung (BioStoffV). Der bei der Bundesanstalt für Arbeitsschutz und Arbeitsmedizin (BAuA) eingerichtete Ausschuss für Biologische Arbeitsstoffe (ABAS) hat SARS-CoV-2 am 19. Februar 2020 vorläufig in die Risikogruppe 3 nach der BioStoffV eingeordnet (zweithöchste Stufe). Grundsätzlich erfolgt die Einstufung in Risikogruppen in den Technischen Regeln für biologische Arbeitsstoffe (TRBA), die von der BAuA veröffentlicht werden, für Viren ist dies die TRBA 462: Einstufung von Viren in Risikogruppen. Beim Auftreten neuartiger, noch nicht zugeordneter Krankheitserreger erfolgt zunächst eine vorläufige Einstufung durch den ABAS. In der Begründung wird auf die Ähnlichkeit von SARS-CoV-2 mit dem SARS-CoV-1 hingewiesen, der die SARS-Pandemie 2002/2003 ausgelöst hat, und auch die Ähnlichkeit in geringerem Umfang mit dem MERS-CoV wird erwähnt. Diese beiden Viren wurden ebenfalls der Risikogruppe 3 zugeordnet. Der ABAS nennt die „derzeit fehlenden Möglichkeiten zu Impfprävention und Therapie sowie die große Verbreitungsmöglichkeit in der Bevölkerung“ als Begründung für die vorläufige Zuordnung zur Risikogruppe 3.
Außerdem werden Empfehlungen zur Arbeit mit dem Virus bei der Diagnostik im Labor gegeben: Nicht gezielte Tätigkeiten (vergleiche § 5 BioStoffV) – ausgehend vom Untersuchungsmaterial, also beispielsweise die Probenvorbereitung, Probenaufbereitung und die Inaktivierung, um den Nachweis mittels RT-PCR (siehe Abschnitt Nachweismethoden) durchzuführen – können unter den Bedingungen der Schutzstufe 2 durchgeführt werden. Dabei sind alle Tätigkeiten, bei denen mit Aerosolbildung zu rechnen ist, in einer mikrobiologischen Sicherheitswerkbank der Klasse II durchzuführen. Außerdem ist die entsprechende persönliche Schutzausrüstung zu tragen. Gezielte Tätigkeiten nach § 5 BioStoffV dürfen nur in Laboratorien der Schutzstufe 3 durchgeführt werden, dies betrifft z. B. die Vermehrung des Virus in einer Zellkultur. Die amerikanische Gesundheitsbehörde CDC hatte zuvor ähnliche Empfehlungen herausgegeben.
Nachweismethoden
Vorgehensweise beim Nachweis
Direkter Nachweis der Viren SARS-CoV-2
RT-PCR-Test
Der sogenannte „PCR-Test“ (genauer: real-time quantitative Reverse-Transkriptase-Polymerase-Kettenreaktion) gilt als Goldstandard für den Nachweis von SARS-CoV-2, da er besonders sensitiv und wenig fehleranfällig ist. Er wird von geschultem Personal in der Regel per Rachenabstrich durchgeführt und im Labor innerhalb weniger Stunden bis Tage ausgewertet.
Quantitative Bestimmung infektiöser Viren
Im Gegensatz zu qualitativen Nachweisen können mit quantitativen Bestimmungen die Mengen aktiver Viren festgestellt werden.
RT-qPCR-Assays messen keine Virustiter infektiöser Viren alleine, das Ergebnis eines PCR-Tests umfasst sowohl infektiöse Viren als auch „ineffizient zusammengesetzte Viren“, inaktive Viren und Virenbruchstücke.
Goldstandardtest zur Quantifizierung infektiöser Viren in einer Probe ist der Plaque-Assay-Test, der nur aktive Viren detektiert. Zur schnellen Messung eines aktiven Virustiters binnen 24 Stunden wäre auch ein Immuno-Plaque-Assay (iPA), eine Kombination aus Plaque-Assay und Immunfluoreszenz-Techniken, geeignet.
Indirekter Nachweis über Antigene und Antikörper
Antigen-Schnelltest

Mit einem Schnelltest (SARS-CoV-2-Antigentest) können innerhalb von knapp 15 Minuten Antigene von SARS-CoV-2 nachgewiesen werden. Er wird durch einen Nasenabstrich oder über eine Speichelprobe mittels eines Lateral-Flow-Tests durchgeführt. Der Antigen-Schnelltest ist nicht so sensitiv wie ein PCR-Test und damit weniger aussagekräftig. Durch das schnellere Ergebnis, die geringeren Kosten und weil er als „Selbsttest“ auch von Laien durchgeführt werden kann, kam ihm in der COVID-19-Pandemie dennoch eine wichtige Rolle zu. Ein positives Testergebnis (angezeigt durch einen – auch nur leicht sichtbaren – zweiten Streifen auf dem Testkit) sollte aber immer durch einen PCR-Test abgesichert werden.
Die Hersteller können ihre COVID-19-Antigen-Schnelltests bis 2022 ohne unabhängige Überprüfung selbst zertifizieren. Das Paul-Ehrlich-Institut stellte im November 2021 eine Studie vor, in der 122 dieser Tests auf Sensitivität untersucht wurden, also auf ihre Fähigkeit, das SARS-CoV-2-Virus nachzuweisen. 96 Tests erfüllten die geforderten Kriterien, 26 hingegen nicht. Ab Mai 2022 müssen die Hersteller zur Zertifizierung ein EU-Referenzlabor hinzuziehen.
Antikörpernachweis

Während die beiden obengenannten Verfahren eine Infektion mit SARS-CoV-2 nachweisen können, wird eine mögliche Immunität durch den Test auf Antikörper überprüft. Dieser erfolgt durch eine Blutprobe, welche ebenfalls mittels eines Schnelltests untersucht werden kann.
Erkrankung COVID-19
Die Infektion mit SARS-CoV-2 kann zur Erkrankung COVID-19 führen. Diese weist ein breites, unspezifisches Symptomspektrum auf, bei leichteren Verläufen oft ähnlich einem grippalen Infekt.
Vorbeugung
Die wichtigsten persönlichen Hygienemaßnahmen zur Vorbeugung sind:
- Persönliche Händehygiene (regelmäßiges Händewaschen mit Seife, mindestens 20 Sekunden lang)
- Augen, Nase oder Mund nicht mit ungewaschenen Händen berühren
- Einhalten des Mindestabstands (1,5 bis 2 Meter) zu anderen Personen außer solchen desselben Haushalts
- Husten oder Niesen nur in Taschentuch oder Armbeuge, keinesfalls in die Hand
- Tragen einer medizinischen Mund-Nasen-Bedeckung (partikelfilternde Halbmaske, FFP2) in öffentlichen Verkehrsmitteln und Gebäuden, insbesondere Spitälern, Heimen und anderen Gemeinschaftseinrichtungen, sowie im Freien, wenn nicht ausreichend Abstand eingehalten werden kann
- Geschlossene Räume ausreichend und häufig lüften
- Die Raumluftqualität kann hierzu mit Hilfe von Kohlenstoffdioxidmessungen überwacht werden, wie zum Beispiel mit CO2-Messgeräten.
- Die Raumluft kann mit Schwebstofffiltern gereinigt werden, um Viren zu entfernen.
- Bei Krankheitsgefühl statt Arztbesuch das Info-Telefon anrufen und zu Hause bleiben
Impfstoffe / Impfung gegen COVID-19
Bei oder nach der klinischen Prüfung der Arzneimittelstudie (Phase-III-Studie) sind unter anderem die RNA-Impfstoffe Tozinameran (BioNTech / Pfizer) und mRNA-1273 (Moderna) sowie die Vektorimpfstoffe Vaxzevria, zuvor AZD1222, (AstraZeneca / Oxford) und Ad26.COV2.S (Janssen / Johnson & Johnson) zugelassen worden. Mit Stand vom 26. März 2021 wurden 278 Projekte vorangetrieben, bei denen neben Vektorimpfstoffen und RNA-Impfstoffen auch DNA-Impfstoffe und klassische Totimpfstoffe gegen COVID-19 entwickelt werden.
Die Anfang des Jahres 2022 dominierende Omikron-Subvariante BA.1 wurde im März 2022 weltweit von der Omikron-Subvariante BA.2 verdrängt. Ab Ende Juni 2022 dominierte dann die Omikron-Subvariante BA.4 und BA.5 das Infektionsgeschehen weltweit. Daher war es notwendig die bereits zugelassenen COVID-19-Impfstoffe rechtzeitig vor einer im Herbst/Winter zu erwartenden Infektionswelle an die Subvarianten BA.4 / BA.5 der Omikronvariante von SARS-CoV-2 anzupassen und rechtzeitig ausreichend Impfstoffdosen für die Auffrischungsimpfung von Personen, die bereits eine Grundimmunisierung gegen COVID-19 erhalten haben, bereit zu stellen.
Am 2. September 2022 sprach die US-Gesundheitsbehörde Zentren für Krankheitskontrolle und -prävention (CDC) eine offizielle Empfehlung für Auffrischungsimpfungen nach abgeschlossener Grundimmunisierung gegen COVID-19 mit einem der beiden bivalenten, an die Omikron-Subvariante BA.4 und BA.5 angepassten mRNA-Impfstoffe Moderna-COVID-19 Vaccine, Bivalent und Pfizer-BioNTech COVID-19 Vaccine, Bivalent aus. Der Beratende Ausschuss für Immunisierungsverfahren des CDC: (ACIP) stimmte mit 13 zu 1 Stimmen für die Auffrischungsimpfungen mit diesen COVID-19-Impfstoffen. Die US-Arzneimittelbehörde (FDA) hatte für diese beiden COVID-19-Impfstoffe bereits am 31. August 2022 eine Notfallzulassung (Emergency use authorization, EUA) für die Verabreichung in den Vereinigten Staaten erteilt. Die Impfstoffe sollen sowohl Schutz vor dem ursprünglichen, erstmals in Wuhan nachgewiesenen Wildtyp von SARS-CoV-2 als auch vor den Omikron-Subvarianten BA.4 und BA.5 bieten. Am 12. September 2022 wurde auch von der Europäischen Kommission auf Empfehlung der Europäischen Arzneimittel-Agentur (EMA) mit Comirnaty Original/Omicron BA.4-5 ein an die Omikron-Varianten BA.4 und BA.5 angepasster COVID-19-Impfstoff für Personen ab 12 Jahren zugelassen, wenn diese bereits eine Grundimmunisierung gegen COVID-19 erhalten haben.
Impfung gegen andere Infektionen
Die Berliner Senatsgesundheitsverwaltung empfahl Ende Februar 2020 allen Menschen über 60 Jahre und chronisch Kranken, ihren Impfstatus zu überprüfen und gegebenenfalls die Impfung gegen Pneumokokken (Impfstoffe wie Pneumovax 23 waren jedoch im März 2020 nur noch eingeschränkt lieferbar) und Keuchhusten (Pertussis) durchführen oder auffrischen zu lassen. Da Menschen über 60 Jahren und chronisch Kranke durch SARS-CoV besonders gefährdet sind, seien sie vorsorglich zu schützen.
Corona-Pandemie
SARS-CoV-2 verursacht die Erkrankung COVID-19 (für englisch corona virus disease 2019), die im Dezember 2019 in der Millionenstadt Wuhan der chinesischen Provinz Hubei auffällig wurde, sich im Januar 2020 in der Volksrepublik China zur Epidemie entwickelte und sich dann weltweit als COVID-19-Pandemie ausbreitete. Um einer Ausbreitung in Staaten ohne leistungsfähige Gesundheitssysteme entgegenzuwirken, rief die Weltgesundheitsorganisation (WHO) am 30. Januar 2020 die Gesundheitliche Notlage internationaler Tragweite (internationale Gesundheitsnotlage) aus. Am 11. März 2020 stufte die WHO die bisherige Epidemie zu einer Pandemie hoch.
Meldepflicht
Literarisches
- Wolfgang Luef: Im Museum gewesen. Überall Corona gesehen. – Klassische Kunst neu interpretiert. Yes Publishing–Pascale Breitenstein GbR, München 2021, ISBN 978-3-96905-105-4.
Dokumentationen
- Brisante Spurensuche – Woher kam das Coronavirus wirklich? Originaltitel: Did Covid Leak from a Lab in China? GB 2021 für Channel 4- television; deutsche Synchronfassung: auf n-TV, 2022; Mitwirkend: Jane Metzler (Autor und Berater des nationalen Sicherheitsrates der USA und der WHO), Alina Chan (Molekularbiologin − The Broad Institute of MIT und Harvard), Nikolai Petrowsky (Mediziner an der Flinders University), David Relman (Mediziner an der Stanford University), Sir John Bell (Mediziner an der University of Oxford), Milton Leitenberg (Zentrum für sicherheitstechnische Studien, University of Maryland), Richard Ebright (Direktor des Mikrobiologischen Instituts der Rutgers University), Gilles Demaneuf (Datenanalyst), Rossana Segreto (Molekularbiologin – Expertin für Genomanalyse), Monali Rahalkar (Mikrobiologin am Agharkar Research Institute [ARI]), Nicholas Wade (Wissenschaftsjournalist bei New York Times und Nature) (auch verfügbar auf youtube.com).
Weblinks
- Nextstrain – Datenbank der genetischen Varianten von SARS-CoV-2
- CoVariants
- cov-lineages.org
- GISAID – Tracking of Variants
Von internationalen Organisationen
- Weltgesundheitsorganisation (WHO): Coronavirus disease (COVID-19) outbreak. In: who.int. (englisch, wird laufend aktualisiert).
- Tracking SARS-CoV-2 variants. (Last Update: 12 October 2021). In: WHO-Website »Home/Activities/Tracking SARS-CoV-2 variants«. Weltgesundheitsorganisation (WHO), 12. Oktober 2021, abgerufen am 8. Januar 2022 (englisch).
Von Behörden in Deutschland
- SARS-CoV-2: Virologische Basisdaten sowie Virusvarianten. In: Website des Robert Koch-Instituts (RKI). Robert Koch-Institut, 30. Juli 2021, abgerufen am 4. September 2021.
- COVID-19 (Coronavirus SARS-CoV-2). Übersichtsseite des Robert Koch-Instituts (RKI). In: rki.de. Robert Koch-Institut, 15. Juni 2021 (wird laufend aktualisiert).
- Antworten auf häufig gestellte Fragen zum Coronavirus SARS-CoV-2. In: rki.de. Robert Koch-Institut, 13. August 2021, abgerufen am 4. September 2021.
Von Behörden in Österreich
- Neuartiges Coronavirus (COVID-19). In: sozialministerium.at, Infektionskrankheiten A-Z. Bundesministerium für Soziales, Gesundheit, Pflege und Konsumentenschutz (Österreich), 23. Dezember 2020, abgerufen am 4. September 2021.
- Informationen zum Coronavirus. In: sozialministerium.at. Bundesministerium für Soziales, Gesundheit, Pflege und Konsumentenschutz (Österreich), 1. September 2021 (wird laufend aktualisiert).
Von Behörden in der Schweiz
- Neues Coronavirus. In: bag.admin.ch. Bundesamt für Gesundheit (Schweiz), 1. September 2021 (wird laufend aktualisiert).
Von anderen Anbietern
- Coronavirus-Pandemie in Deutschland: Herausforderungen und Interventionsmöglichkeiten. (PDF; 158 kB) Nationale Akademie der Wissenschaften Leopoldina, 21. März 2020.
- Ewen Callaway: The coronavirus is mutating – does it matter? In: Nature – a weekly journal of science. Nature – About the Journal, 16. September 2020, abgerufen am 24. Dezember 2020 (englisch, doi:10.1038/d41586-020-02544-6).
- Corona in 3D: Ein Blick ins Innere des Virus. (Interaktive 3D-Animation, Quelle: dpa, AFP, Reuters, ZDF). In: ZDFheute. 8. April 2021, abgerufen am 17. April 2021 (Bekanntes und weniger Bekanntes über das neuartige Coronavirus).
- SARS-CoV-2-Varianten: Evolution im Zeitraffer. In: aerzteblatt.de (Dtsch Arztebl 2021; 118(9): A-460 / B-388). Hrsg.: Bundesärztekammer und Kassenärztliche Bundesvereinigung, abgerufen am 14. Mai 2021.
- Beweglicher als gedacht – neue Erkenntnisse über das Spikeprotein von SARS-CoV-2. In: PEI-Pressemitteilung 14 / 2020. Paul-Ehrlich-Institut (PEI), 18. August 2020, abgerufen am 14. Mai 2021 (Videosimulation von vier Spike-Proteinen des SARS-CoV-2-Virus vom Max-Planck-Institut für Biophysik).
- Kai Kupferschmidt in Science: Evolving threat – New variants have changed the face of the pandemic. What will the virus do next? science.org, 19. August 2021, abgerufen am 30. August 2021 (englisch, Version in Science, Vol 373, Issue 6557 – Ausblick weitere Entwicklung).
- Bas B. Oude Munnink et al.: The next phase of SARS-CoV-2 surveillance: real-time molecular epidemiology. In: Nature Medicine, review articles. nature.com, 9. September 2021, abgerufen am 13. September 2021 (englisch, PMID 34504335 / Gesamtüberblick SARS-CoV-2 September 2021).
- Thomas Böldicke: Varianten von SARS-CoV-2: Wie viel Schutz noch bleibt. Medizinreport. In: Deutsches Ärzteblatt 118(38): A-1701 / B-1410. 2021, abgerufen am 17. August 2022 („Deutschland ist in der 4. Pandemiewelle angekommen. Virusvarianten erschweren die Bekämpfung von SARS-CoV-2. Neue Studien zeigen, inwiefern frühere Infektionen sowie die verfügbaren Coronaimpfstoffe vor neuen Infektionen und schweren Krankheitsverläufen schützen.“).
Anmerkungen
–