
ACE-Hemmer (kurz für Angiotensin-Converting-Enzyme-Hemmer) sind gefäßerweiternde, damit den Gefäßwiderstand senkende, und die Freisetzung der blutdrucksteigernden Katecholamine Noradrenalin und Adrenalin hemmende Arzneistoffe, die insbesondere in der Therapie des Bluthochdruckes (arterielle Hypertonie) und der chronischen Herzinsuffizienz Anwendung finden. Sie sind Hemmstoffe (Inhibitoren) des Angiotensin-konvertierenden Enzyms (Angiotensin Converting Enzyme), das im Wesentlichen die Umwandlung von inaktivem Angiotensin-I in aktives Angiotensin-II bewirkt und ein Teil einer den Blutdruck steuernden Kaskade ist (Renin-Angiotensin-Aldosteron-System). ACE-hemmende Inhaltsstoffe wurden zuerst in Schlangengiften gefunden. Die wichtigsten in der Therapie verwendeten Wirkstoffe dieser Kategorie sind Captopril, Enalapril, Lisinopril, Perindopril und Ramipril.
Chemie


ACE-Hemmer, wie Captopril, Enalapril und ihre Nachfolgersubstanzen, sind strukturverwandt mit dem aus dem Schlangengift der brasilianischen Jararaca-Lanzenotter (Bothrops jararaca) isolierten Pentapeptid BPP5a (von „Bradykinin potenzierendes Peptid“; Sequenz DKWAP, siehe Abbildung). Die in BPP5a vorkommende Tripeptidsequenz Tryptophan-Alanin-Prolin wurde als wirksame Komponente erkannt (in Abbildung rot dargestellt).
Da BPP5a und das Tripeptid im Körper sehr schnell abgebaut werden, wurden zahlreiche Modifikationen am Molekül vorgenommen, um die Wirkdauer zu verlängern. Dazu wurde die WAP-Sequenz gegen eine ähnliche aber stabilere FAP-Sequenz ausgetauscht. Die Einbringung einer bernsteinsäure- oder glutarsäureanalogen Struktur (in Abbildung grün dargestellt) brachte weitere Stabilität und eine Verstärkung der Hemmwirkung am Angiotensin Converting Enzyme.
Darüber hinaus sind bis auf Captopril und Lisinopril alle therapeutisch genutzten ACE-Hemmer Prodrugs, die erst im Körper aktiviert werden. Im Falle von Enalapril und Ramipril geschieht dies durch Abspaltung der Ethylgruppe durch Esterasen, wodurch die Wirkform, das Enalaprilat bzw. Ramiprilat, mit einer freien Carboxygruppe entsteht, die das Zink des ACE komplexieren kann.
Pharmakologie
Anwendungsgebiete
ACE-Hemmer werden überwiegend zur Therapie des Bluthochdrucks eingesetzt. Hierfür gelten sie einzeln (Monotherapie) und in Kombination mit anderen Blutdrucksenkern (Kombinationstherapie, insbesondere mit Diuretika oder Calciumantagonisten) als Mittel der ersten Wahl. Bei Bluthochdruckformen, die mit einem erniedrigten Renin-Spiegel im Blutplasma einhergehen (z. B. Conn-Syndrom) zeigen ACE-Hemmer hingegen nur unzureichende Wirksamkeit.
Daneben haben sich einige ACE-Hemmer in zahlreichen großen klinischen Studien auch bei der chronischen Herzinsuffizienz als lebensverlängernd erwiesen. Dies beruht wahrscheinlich auf der Senkung der Nachlast und Verminderung der Wandspannung des Herzmuskels durch die Abnahme von Angiotensin II.
Auch nach Herzinfarkten und bei einer Herzmuskelentzündung werden ACE-Hemmer eingesetzt.
Eine weitere Indikation der ACE-Hemmer ist die diabetische Nephropathie. Die kanadische ONTARGET Studie weist aber darauf hin, dass ACE-Hemmer auf keinen Fall in Kombination mit Angiotensin-II-Rezeptorblockern eingenommen werden dürfen. Während beide Medikamente für sich genommen nephroprotektiv wirken, kam es bei der Kombinationstherapie zu einer signifikant verschlechterten Nierenfunktion. Ferner zeichnete sich ein Trend in Richtung eines Anstiegs der Dialysepflicht ab. Untersucht wurden u. a. Ramipril und Telmisartan.
Wirkmechanismus

Der Wirkungsmechanismus der ACE-Hemmer beruht auf einer Hemmung des Angiotensin-I-umsetzenden Enzyms ACE. Dieses Enzym hat im Organismus zwei Hauptaufgaben: Einerseits ist es für die Synthese des gefäßverengend wirksamen Octapeptids (Peptid aus acht Aminosäuren) Angiotensin II aus seiner inaktiven Vorstufe, dem Decapeptid (zehn Aminosäuren) Angiotensin I unter Abspaltung der zwei C-terminalen Aminosäuren zuständig. Andererseits katalysiert es den Abbau des Mediators Bradykinin in inaktive Produkte.
Die Hemmung des Angiotensin Converting Enzyme hat eine Abnahme der Angiotensin-II-Konzentration an den Angiotensinrezeptoren (AT1 und AT2) zur Folge. Primär sinkt dadurch der Blutgefäßtonus, und der Blutdruck nimmt ab. Es kommt also hämodynamisch zu einer Senkung der Vorlast und der Nachlast. Sekundär führt die Abnahme des Angiotensin-II-Spiegels zu einer Verringerung der Aldosteron-Freisetzung aus der Nebennierenrinde und somit zu einer Beeinflussung des Wasserhaushalts (siehe auch Renin-Angiotensin-Aldosteron-System, RAAS). Auf zellulärer Ebene kann ein Rückgang der durch Angiotensin II vermittelten mitogenen Effekte an Fibroblasten und Myozyten des Herzens, die insbesondere nach einem Herzinfarkt zu ungünstigen Veränderungen (Remodeling) führen, beobachtet werden. In welchem Ausmaß die Blutdrucksenkung erfolgt, hängt davon ab, wie hoch die Aktivität des RAA-Systems ist. Bei der Herzinsuffizienz ist die Aktivität des RAAS sehr hoch, sodass man auf jeden Fall nur einschleichend dosieren sollte, um eine zu starke Blutdrucksenkung zu vermeiden.
Bei Nierenerkrankungen wie der diabetischen Nephropathie führen ACE-Hemmer zu einer verminderten Proteinausscheidung und verhindern ein Fortschreiten der Erkrankung (Nephroprotektion).
Die Hemmung des Abbaus von Bradykinin führt hingegen zu dessen Kumulation und damit verbundenen Nebenwirkungen.
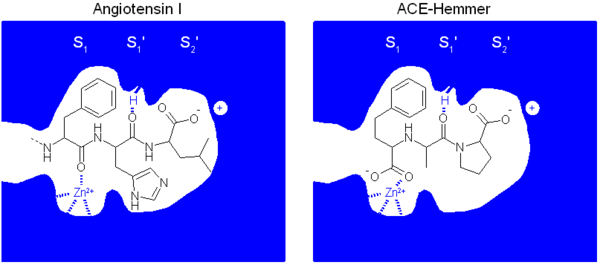
Molekularer Wirkmechanismus
Auch der molekulare Wirkmechanismus der ACE-Hemmer konnte aufgeklärt werden. Er beruht auf der Ähnlichkeit der ACE-Hemmer zu einem Peptidkettenende des Angiotensin I. Dadurch werden ACE-Hemmer vom Angiotensin Converting Enzyme fälschlich für das physiologische Substrat Angiotensin I gehalten. Im Gegensatz zum physiologischen Substrat werden sie aber nicht vom Enzym umgesetzt, sondern blockieren es. Wichtig für die Bindung des Liganden sind drei Wechselwirkungen:
- eine Komplexierung des Zink-Ions des ACE. Dies ist in der Regel eine Carboxygruppe oder beim Captopril eine Thiolgruppe
- eine elektrostatische Wechselwirkung zwischen dem K511 des ACE und der Carboxylatfunktion des Prolins des Liganden
- eine Wasserstoffbrückenbindung zwischen H353 des ACE und dem Carbonyl des Alanins bzw. Lysins des Liganden
Pharmakokinetik
Entsprechend ihrer chemischen Differenzen unterscheiden sich die ACE-Hemmer in ihrer Pharmakokinetik. Die Mehrzahl der derzeit verfügbaren ACE-Hemmer sind Prodrugs. Das heißt, dass sie nach einer 20%igen (Ramipril) bis fast 100%igen Aufnahme (Resorption) durch Enzyme im Körper aktiviert werden müssen (siehe Chemie). Lediglich Captopril und Lisinopril benötigen diesen Aktivierungsschritt nicht. Maximale Plasmaspiegel der Wirkformen werden nach 1 bis 8 Stunden erreicht. Die Plasmahalbwertszeiten schwanken zwischen 2 (Captopril) und 40 Stunden (Spirapril). Entsprechend variiert auch die Wirkdauer (8 bis 48 Stunden). Alle ACE-Hemmer werden überwiegend über die Niere ausgeschieden. Fosinopril, Moexipril und Spirapril zeigen darüber hinaus eine relevante biliäre Exkretion (Ausscheidung über die Galle).
Nebenwirkungen
Die wichtigsten Nebenwirkungen sind trockener Husten, Hypotonie, akutes Nierenversagen, Hyperkaliämie und Probleme während der Schwangerschaft (unten einzeln erklärt). Diese Nebenwirkungen sind allen ACE-Hemmern gemeinsam. Bei einer 2018 veröffentlichten Kohortenstudie mit fast einer Million Patienten war die Verwendung von ACE-Hemmern, nach fünf Jahren der Anwendung, mit einem insgesamt um 14 % erhöhten Risiko für Lungenkrebs verbunden. Bei Patienten, die ACE-Hemmer mehr als zehn Jahre lang verwendeten, lag ein um 31 % erhöhtes Risiko vor.
Die meisten Nebenwirkungen von ACE-Hemmern werden mit einem verlangsamten Abbau und einer Anreicherung von Bradykinin durch ACE-Hemmer in Verbindung gebracht. Dazu zählen Hautreaktionen wie z. B. Exantheme (0,1–1 %) und Nesselsucht (0,01–0,1 %). Schwere allergische Hautreaktionen werden hingegen nur sehr selten beobachtet (< 0,01 %). Die als charakteristisch für ACE-Hemmer geltende Nebenwirkung, das Auftreten von Angioödemen, kann ebenfalls nur selten beobachtet werden (0,01–0,1 %).
Auch die Mehrzahl der die Atemwege betreffenden Nebenwirkungen kann mit einer Kumulation von Bradykinin in Verbindung gebracht werden. Dazu zählt in erster Linie ein trockener Husten, der in den ersten drei Monaten bei 5–35 % der Patienten auftritt. Diese Nebenwirkung ist nicht dosisabhängig. Bei trockenem Husten sollte der ACE-Hemmer abgesetzt bzw. gegen ein anderes Medikament entsprechend der Indikation ausgetauscht werden.
Auch Heiserkeit und Halsschmerz (0,1–1 %) treten auf. Asthmaanfälle und Atemnot können ebenfalls, wenn auch selten, auftreten (0,01–0,1 %).
Unter der Therapie mit ACE-Hemmern kann es bradykininunabhängig zu einer Hypotonie, d. h. zu einer zu starken Blutdrucksenkung kommen. Infolgedessen können gelegentlich Schwindel, Kopfschmerz und Benommenheit beobachtet werden (0,1–1 %). Von schweren Herz-Kreislauf-Ereignissen, wie Angina Pectoris, Herzinfarkt und Synkope, wurde nur in Einzelfällen berichtet. Dieser Nebenwirkung (die bevorzugt bei Patienten mit Herzinsuffizienz auftritt) kann man mit Vorsichtsmaßnahmen vorbeugen: bei Flüssigkeitsmangel zuerst Flüssigkeitsgabe und Absetzen von Diuretika (falls der Patient diese einnimmt), dann mit der Einnahme von ACE-Hemmern beginnen; bei Patienten mit Herzinsuffizienz mit einer geringeren Dosierung als anvisiert anfangen, dann die Dosis steigern.
Durch Eingriff in den Wasser- und Elektrolythaushalt können gelegentlich funktionelle Nierenfunktionsstörungen beobachtet werden (0,1–1 %). Eine Proteinurie (vermehrte Ausscheidung von Proteinen im Harn) bis hin zu akutem Nierenversagen wurde hingegen nur selten beobachtet (0,01–0,1 %). Zum akuten Nierenversagen kommt es fast nur bei Risikopatienten, d. h. bei Menschen oder Tieren mit bilateraler Nierenarterienstenose, mit einer hypertonischen Nephrosklerose, mit einer Herzinsuffizienz, mit einer polyzystischen Nierenerkrankung oder mit einem vorher existierenden chronischen Nierenversagen. Das Nierenversagen ist oft reversibel.
Eine klinisch relevante Hyperkaliämie kommt bei < 10 % vor, bei fast allen Patienten kann eine geringe, klinisch nicht relevante Erhöhung des Kalium-Spiegels beobachtet werden. Die klinisch relevante Hyperkaliämie entsteht sehr oft bei Patienten mit bereits vorhandenem Nierenversagen, gleichzeitiger Einnahme von kaliumsparenden Diuretika (z. B. Triamteren), NSAR (nichtsteroidale Antirheumatika), schwerer Herzinsuffizienz und bei älteren Patienten. Niedrige Dosen von ACE-Hemmern führen nicht zu dieser Nebenwirkung.
Durch die Wirkungen auf das Renin-Angiotensin-Aldosteron-System mit Abnahme der Aldosteron-Ausschüttung lässt sich diese weitere unerwünschte Wirkung von ACE-Hemmern erklären: Aldosteron verstärkt die Natrium- und Wasser-Wiederaufnahme in der Niere, während es die Kalium-Ausscheidung fördert. Bei verminderter Konzentration von Aldosteron kommt es zum gegenteiligen Effekt: erhöhte Natrium- und Wasser-Ausscheidung der Niere, während Kalium vermehrt im Körper verbleibt. So kann es zu einer vor allem für das Herz gefährlichen Hyperkaliämie kommen. Selten kommt es auch zu einer Hyponatriämie.
Kontraindiziert in der Schwangerschaft: Da ACE-Hemmer in der Schwangerschaft u. a. Wachstums- und Knochenbildungsstörungen beim Kind, verbunden mit einer erhöhten Sterblichkeit hervorrufen können, dürfen ACE-Hemmer in dieser Zeit nicht eingenommen werden und sollten durch andere therapeutische Maßnahmen ersetzt werden. Siehe z. B. die Aplasia cutis congenita.
Wechselwirkungen
ACE-Hemmer verstärken die Blutbild verändernden Nebenwirkungen immunsuppressiv wirkender Arzneistoffe (Immunsuppressiva, Zytostatika und Glucocorticoid). Ebenso verstärken ACE-Hemmer die Blutzucker senkende Wirkung oraler Antidiabetika und von Insulin.
Durch Eingriff in den Wasser- und Elektrolythaushalt kann die Ausscheidung von Lithium verlangsamt werden. Ebenso kann eine Verstärkung des Anstiegs des Kaliumspiegels bei kombinierter Anwendung mit kaliumsparenden Diuretika beobachtet werden.
Bei Kombination mit anderen blutdrucksenkenden Arzneimitteln sollte eine verstärkte Blutdrucksenkung berücksichtigt werden. Synergistische Effekte, die auch therapeutisch ausgenutzt werden, treten insbesondere mit Diuretika und mit Calciumkanalhemmern auf. Eine verringerte blutdrucksenkende Wirkung der ACE-Hemmer konnte vereinzelt nach Einnahme kochsalzreicher Kost beobachtet werden.
Arzneistoffe
Die internationalen Freinamen der einzelnen ACE-Hemmer enden auf -pril. Derzeit sind in Deutschland folgende ACE-Hemmer als Arzneistoff (Substanz bzw. Prodrug) zugelassen:
- Benazepril
- Captopril
- Cilazapril (Wirkmetabolit: Cilazaprilat)
- Enalapril
- Fosinopril
- Imidapril
- Lisinopril
- Moexipril
- Perindopril (Wirkmetabolit: Perindoprilat)
- Quinapril (Wirkmetabolit: Quinaprilat)
- Ramipril (Wirkmetabolit: Ramiprilat)
- Spirapril
- Trandolapril
- Zofenopril
Geschichte
Seit 1980 spielen die ACE-Hemmer eine wichtige Rolle für die Behandlung von koronaren Herzkrankheiten. Dafür mussten zuerst Erkenntnisse über das Renin-Angiotensin-Aldosteron-System (RAAS) gewonnen werden. Dessen Erforschung startete 1898 durch die Isolierung des Renins, ermöglicht durch Robert Tigerstedt und den Medizinstudenten Per Gustav Bergman. Harry Goldblatt postulierte die Beteiligung des Enzyms an der Blutdruckregulation. Diese Hypothese konnte allerdings erst 1939 bewiesen werden. 1946 folgten dann Berichte, die aufzeigten, dass Patienten mit chronischer Herzinsuffizienz eine erhöhte Reninaktivität aufweisen. Aus diesem Grund verstärkte man ab den 1950er Jahren die Erforschung des RAAS bei Hypertonie.
Der Grundstein für die Entwicklung der ACE-Hemmer wurde 1956 mit der Aufklärung der Funktion des Angiotensin Converting Enzyme (ACE) durch Leonard T. Skeggs und Joe Kahn gelegt. Die Bedeutung dieses Enzyms für die Blutdruckregulation wurde anfangs noch unterschätzt.
14 Jahre nach der Entdeckung des Angiotensin Converting Enzyme fand der Pharmakologe Sérgio Henrique Ferreira 1965 heraus, dass das Gift der Jararaca-Lanzenotter in vitro zu einer Hemmung dieses Enzyms führt. 1970 isolierten er sowie, unabhängig von ihm, Miguel Ondetti das Pentapeptid BPP5a aus dem Schlangengift, welches die Angiotensin-I-Konversion hochspezifisch hemmt.
Da BPP5a im Körper sehr instabil ist, startete fast gleichzeitig eine Suche nach potenteren und stabileren Inhibitoren des Enzyms. Ein erster Erfolg gelang 1971 mit der Entdeckung der ACE-hemmenden Wirkung des Nonapeptids Teprotid. Die Hersteller stellten die klinische Weiterentwicklung von Teprotid jedoch zwei Jahre später wegen mangelnden kommerziellen Interesses ein. Zudem musste Teprotid intravenös verabreicht werden, wodurch es sich für chronische Erkrankungen wie die Hypertonie als ungeeignet erwies.
Die erste Synthese eines oralen ACE-Hemmers gelang David Cushman und Ondetti: Mithilfe des Wissens über die Strukturähnlichkeit des ACE mit der im Pankreas vorkommende Carboxypeptidase A konnte die Verbindung Succinylprolin hergestellt werden, welche bei weitem nicht so wirksam wie Teprotid war.
Anfangs der 1970er-Jahre konnte die wirksame Teilstruktur der ACE-hemmenden Peptide BPP5a und Teprotid aufgeklärt werden. Aufgrund dieser Entdeckungen wurden neue nichtpeptidische ACE-Hemmer entwickelt. 1974 wurde erstmals der ACE-Hemmer Captopril als Produkt einer groß angelegten Wirkstoffsuche (Screening) der Pharmafirma Squibb beschrieben. 1981 wurde er als erster ACE-Hemmer unter dem Handelsnamen Lopirin in die Therapie eingeführt. Captopril ist in der Wirkstärke mit der von Teprotid gleichzusetzen.
In den folgenden Jahren versuchte man strukturähnliche Verbindungen zu Captopril zu entwickeln. Dadurch stieß man auf Verbindungen mit SH-Gruppen, welche eine höhere Lipophilie besitzen. Da Captopril anfangs bei klinischen Studien in relativ hohen Dosen verwendet wurde, traten zahlreiche, teilweise auch schwerwiegende Nebenwirkungen auf, die man u. a. dem Sulfyhydrylanteil im Molekül zuschreiben konnte. Aus diesem Grund verringerte man die Dosen, wodurch auch Auftreten der Nebenwirkungen deutlich abnahm. Dennoch bemühte man sich weiter um die Synthese eines ACE-Hemmers ohne Sulfylhydrylanteil. Dieses Ziel wurde 1980 erreicht, indem Arthur A. Patchett und Charles S. Sweet den sulfylhydrylfreien ACE-Hemmer Enalapril bzw. Enalaprilat synthetisierten und vorstellten. Letzteres hatte allerdings nur eine geringe Bioverfügbarkeit, weshalb es in Form des Ethylesters als Prodrug unter den Handelsnamen Pres und Xanef auf den Markt gelangte. Das Enalapril galt nun als „Prototyp“ für andere strukturähnliche ACE-Inhibitoren. Aufgrund des großen therapeutischen und wirtschaftlichen Erfolges der Arzneistoffe Captopril und Enalapril wurde eine zweite Generation der ACE-Hemmer entwickelt, die seit Anfang der 1990er Jahre erhältlich sind.
Ökonomische Bedeutung
In Deutschland nehmen etwa 20 % der Bevölkerung und jeder zweite über 55 Jahre Arzneimittel zur Behandlung des Bluthochdrucks ein. ACE-Hemmer sind mit einem Anteil von über 50 % die meistverordneten Antihypertensiva. Etwa 80 % der mit einem ACE-Hemmer behandelten Bluthochdruckpatienten verwenden ein Monotherapeutikum, der Rest nutzt ein Kombinationspräparat. Die Verordnungszahlen, die in Deutschland im Jahr 2009 etwa 5 Milliarden definierte Tagesdosen (DDD) erreichten, nahmen in den letzten zehn Jahren linear um etwa 200 % zu. Auf dem von Generika geprägten deutschen Markt dominiert der Arzneistoff Ramipril (68 %) deutlich vor Enalapril (18 %) und Lisinopril (10 %).
Alternativen
Neuere Substanzen aus der Gruppe der AT1-Antagonisten (Sartane) hemmen nicht mehr das Angiotensin Converting Enzyme, sondern wirken antagonistisch auf den Angiotensin-II-Rezeptor-1-Subtyp, sodass Nebenwirkungen seltener auftreten. Ihre bessere Verträglichkeit beruht darauf, dass sie nicht auf das Bradykinin-System einwirken. AT1-Antagonisten sind seit einigen Jahren ebenfalls als Generika auf dem Markt (z. B. 2008 Eprosartan, 2010 Losartan), jedoch noch immer teurer als ACE-Hemmer.
Ein anderer Angriffspunkt ist die Hemmung des in der Niere gebildeten Enzyms Renin, das für die Synthese von Angiotensin I verantwortlich ist. Mit Aliskiren ist im Jahr 2007 ein selektiver Hemmer dieses Enzyms zugelassen worden, weitere Reninhemmer wie z. B. Remikiren und Zankiren befinden sich in der klinischen Erprobung.
Vasopeptidaseinhibitoren wie Omapatrilat sind von den klassischen ACE-Hemmern abgeleitet und standen 2010 noch vor der Zulassung durch die Gesundheitsbehörden, da es in Studien zu schweren Angioödemen kam. Zusätzlich zur Hemmung des Angiotensin Converting Enzyme hemmen die Vasopeptidaseinhibitoren die neutrale Endopeptidase, ein Enzym, das für die Inaktivierung des blutgefäßrelaxierenden atrialen natriuretischen Peptids (ANP) verantwortlich ist.
Intensivmedizinischer Aspekt
In der Intensivmedizin hat sich gezeigt, dass Patienten, die vor dem Intensivstationsaufenthalt mit ACE-Hemmern therapiert wurden, oftmals einen höheren Verbrauch an Katecholaminen aufweisen, um den mittleren arteriellen Druck zu stabilisieren. Grund dafür dürfte ein Vasopressinmangel sein, der auf die vorhergehende Therapie mit ACE-Hemmern zurückzuführen wäre. Durch die Substitution von Vasopressin (ADH) kann häufig der Katecholaminbedarf (soweit keine weiteren Gründe für niedrigen Blutdruck vorliegen) rasch reduziert werden und danach das Vasopressin binnen 12–24 Stunden ausgeschlichen werden.
Literatur
- D. W. Cusham, M. A. Ondetti: History of the design of captopril and related inhibitors of angiotensin converting enzyme. In: Hypertension. Baltimore, 17.1991, S. 589–592. PMID 2013486.
- E. Mutschler, G. Geisslinger, H. K. Kroemer, M. Schäfer-Korting: Therapie der Hypertonie. In: E. Mutschler (Hrsg.): Arzneimittelwirkungen. Wissenschaftliche Verlagsgesellschaft, Stuttgart 2001, ISBN 3-8047-1118-9, S. 571–587.