
| Klassifikation nach ICD-10 | |
|---|---|
| E84 | Zystische Fibrose |
| E84.0 | Zystische Fibrose mit Lungenmanifestationen |
| E84.1 | Zystische Fibrose mit Darmmanifestationen |
| E84.8 | Zystische Fibrose mit sonstigen Manifestationen |
| E84.9 | Zystische Fibrose, nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
Mukoviszidose (abgeleitet von lateinisch mucus ‚Schleim‘, und viscidus ‚zäh‘ bzw. ‚klebrig‘), auch zystische Fibrose (englisch cystic fibrosis, CF) genannt, ist eine autosomal-rezessiv vererbte Stoffwechselerkrankung.
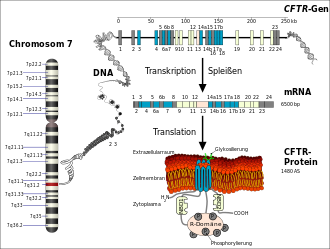
Die Ursache dieser Erkrankung ist eine durch Mutation bedingte Fehlfunktion von Chloridkanälen bestimmter Körperzellen, des Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), wodurch die Zusammensetzung aller Sekrete exokriner Drüsen verändert wird. Die betroffenen Zellen sind nicht in der Lage, mittels Osmose Wasser in das umliegende Gewebe zu ziehen, wodurch der Wassergehalt des Bronchialsekrets sowie der Sekrete der Bauchspeicheldrüse, der Leber (Galle), der inneren Geschlechtsorgane und der akzessorischen Geschlechtsdrüsen sowie des Dünndarms und der Schweißdrüsen zu niedrig ist. Die Sekrete werden dadurch zähflüssig, und in den betroffenen Organen kann es zu Funktionsstörungen unterschiedlicher Art kommen.
Mukoviszidose ist daher eine Multisystemerkrankung. Es sind über 2000 verschiedene Mutationen bekannt, die bei den Betroffenen zu einer unterschiedlichen Ausprägung der Mukoviszidose führen können. Mukoviszidose ist die häufigste autosomal-rezessive Erbkrankheit und die häufigste letale genetische Erkrankung in der hellhäutigen Bevölkerung. Statistisch gesehen kommt in dieser Bevölkerungsgruppe auf etwa 2000 Lebendgeburten ein erkranktes Kind. Dabei gibt es erhebliche regionale Schwankungen in der Häufigkeit der Erkrankung.
Erste Symptome zeigen sich bereits in der frühen Kindheit. Mukoviszidose kann vorgeburtlich diagnostiziert werden. Die tödlich endende Erkrankung ist derzeit nicht heilbar. Durch den medizinischen Fortschritt konnten über die letzten Jahrzehnte neue Behandlungsmöglichkeiten etabliert werden, durch die die mittlere Lebenserwartung auf mittlerweile 57 Jahre erheblich gesteigert werden konnte. Mit neuen, personalisierten Behandlungskonzepten ist zukünftig eine weitere Verbesserung der Lebenserwartung zu erwarten. Die Krankheit ist Gegenstand intensiver Forschungen.
Die beiden Begriffe Mukoviszidose und zystische Fibrose beschreiben unterschiedliche Symptome derselben Krankheit. Im deutschsprachigen Raum wird – im Gegensatz zum englischsprachigen – die Bezeichnung Mukoviszidose bevorzugt.
Epidemiologie
In Europa liegt die Wahrscheinlichkeit der Geburt eines Kindes mit Mukoviszidose bei etwa 1:2.000. In Deutschland leben derzeit rund 8.000 an Mukoviszidose erkrankte Menschen. Weltweit sind es insgesamt etwa 70.000, wovon je 30.000 auf Europa und Nordamerika entfallen. Pro Jahr werden etwa 300 Kinder mit Mukoviszidose in Deutschland geboren. Mukoviszidose stand an erster Stelle der Erbkrankheiten, die früher bei Kindern bis zum 15. Lebensjahr zum Tod führten.
| Mutation | Genabschnitt | Mutationstyp | Klasse | D [%] | A [%] | CH [%] |
| ΔF508 | Exon 10 | Aminosäuredeletion | II | 71,8 | 62,9 | 64,1 |
| R553X | Exon 11 | Stoppmutation | I | 2,0 | 1,7 | 3,5 |
| N1303K | Exon 21 | Aminosäuresubstitution | II | 1,8 | 0,6 | 1,6 |
| R347P | Exon 7 | Aminosäuresubstitution | IV | 1,2 | 1,6 | 0,8 |
| G542X | Exon 11 | Stoppmutation | I | 1,2 | 3,3 | 1,6 |
| G551D | Exon 11 | Aminosäuresubstitution | III | 0,9 | 1,2 | 0,2 |
| 1717 1G→A | Intron 10 | Spleißmutation | I | 0,9 | 0,8 | 3,8 |
| 2789 +5G→A | Intron 14b | Spleißmutation | IV | 0,9 | 2,4 | 0,3 |
| 3905insT | Exon 20 | Frameshift-Mutation | I | - | - | 4,8 |
Die Allelfrequenz liegt in der deutschen Bevölkerung bei etwa 0,02 bis 0,025. Diese Zahl ist ein Maß für die relative Häufigkeit eines Allels in einer Population. Daraus folgt, dass ca. 4 % der Bevölkerung, also jeder Fünfundzwanzigste, ein defektes CFTR-Gen trägt. Diese rund drei Millionen Menschen allein in Deutschland sind (mit Ausnahmen) gesunde Genträger, die das mutierte Allel weitervererben können. Man spricht in diesem Fall von heterozygoten Merkmalsträgern. Die Wahrscheinlichkeit, dass zwei heterozygote Merkmalsträger ein Kind zeugen, hat – bezogen auf die Gesamtpopulation – dann eine Wahrscheinlichkeit von 0,02² = 0,0004. Bei einer Bevölkerung von 81,2 Millionen Menschen (Stand Dezember 2014, statistisches Bundesamt) entspräche dies mathematischen 32.000 Einwohnern. In epidemiologischen Studien wurde für Deutschland ein Wert von 1:3.300 ermittelt. Weltweit betrachtet sind die Mukoviszidose-Inzidenzen sehr unterschiedlich. Die weltweit höchste Wahrscheinlichkeit für die Geburt eines Kindes mit Mukoviszidose hat Irland mit 1:1.800. Den in Europa niedrigsten Wert weist Finnland mit 1:25.000 auf. Bei Menschen afrikanischer Abstammung beträgt das Risiko etwa 1:17.000. Für Menschen asiatischer Abstammung ist es mit etwa 1:100.000 am unwahrscheinlichsten, mit der Erkrankung geboren zu werden. Beispielsweise liegt der Wert in Japan bei 1:350.000.
Juni 2017 sind 2019 unterschiedliche Mutationen im CFTR-Gen statistisch erfasst. Diese Mutationen sind über die Gesamtbevölkerung ungleichmäßig verteilt. So haben weniger als 20 Mutationen einen Anteil von über 0,1 % und nur fünf Mutationen einen Anteil von über 1 % an der Gesamtzahl der Mukoviszidoseerkrankungen. Die mit Abstand am häufigsten auftretende Mutation hat die Bezeichnung ΔF508. Sie findet sich in etwa 2/3 aller CFTR-Allele von Mukoviszidosepatienten. Innerhalb von Europa nimmt dabei die Prävalenz von Nord-West nach Süd-Ost ab.
Bei den 2019 erfassten Mutationen stellen nicht-synonyme Mutationen, das sind Punktmutationen, bei denen für eine andere Aminosäure codiert wird, mit 39,4 % den Hauptanteil. Es folgen Frameshift-Mutationen, das sind Verschiebungen des Leserasters von CFTR auf der DNA, mit 15,7 % und Spleißmutationen mit 11,3 %. Der Anteil an Nonsense-Mutationen liegt bei 8,4 %, der von in frame-Deletionen und in frame-Insertionen bei 2,1 %. Große Insertionen und Deletionen haben eine Häufigkeit von 2,6 %. Promotermutationen im CFTR-Gen, das sind Punktmutationen im Promoterbereich von CFTR, die zu einer verminderten Genexpression von CFTR führen, sind mit einem Anteil von 0,74 % vergleichsweise selten. Lediglich 13,3 % der erfassten Mutationen in codierenden Bereichen von CFTR führen nicht zur Erkrankung. Bei 6,5 % ist der Mutationstyp noch unbekannt.
Durch den Gründereffekt können manche, eher seltene Mutationen in einigen Populationen deutlich überrepräsentiert sein. Der Gründereffekt kann dabei durch religiöse, ethnische oder geografische Isolation entstehen. So tritt die Stoppmutation W1282X beispielsweise bei aschkenasischen Juden, die Deletion 394delTT in nordischen Völkern, die Insertion 3905InsT in der Schweiz, die Aminosäuresubstitution S549R bei Beduinen und die Spleißmutation 3120+1G→A auf dem afrikanischen Kontinent vergleichsweise häufig bei Mukoviszidosepatienten auf. Ein Spezialfall ist die Mutation 3905InsT. Sie ist nur in der Schweiz, bei der Amischen Gemeinde in Nordamerika und bei Akadiern verbreitet. In der Schweiz liegt sie mit einer Häufigkeit von 4,8 % auf dem zweiten Platz der CFTR-Mutationen. Bei den Amischen wird sogar ein Wert von 16,7 % und bei Akadiern von 14,3 % erreicht. Ursache hierfür ist der Gründereffekt von Auswanderern aus der Deutschschweiz, die im 18. Jahrhundert die Amische Gemeinde gründeten und sich auch in Louisiana niederließen. In Deutschland findet sich die ΔF508-Mutation bei 72 % der Patienten, die restlichen 28 % weisen allerdings ein ausgesprochen heterogenes und vielseitiges Spektrum auf, was die genetische Diagnostik erschwert. Über 80 unterschiedliche Mutationen wurden bisher bei deutschen Mukoviszidosepatienten nachgewiesen. Da die Mutationsform den Krankheitsverlauf und zunehmend auch die Behandlungsmöglichkeiten beeinflusst, lässt sich die klinische Prognose bei vergleichsweise seltenen Mutationsformen im CFTR-Gen häufig nur schwer stellen. Es gibt zu wenige Patienten und somit kaum Fallbeispiele mit demselben CFTR-Genotyp. Von den 1995 bekannten Mutationen ist etwa jede Dritte so selten, dass sie bisher nur in einer einzigen Familie gefunden wurde (engl. private mutation). Spontanmutationen sind ausgesprochen selten. Weltweit wurden bisher erst vier Fälle beschrieben.
Genetik und Molekularbiologie
Die Ursache für Mukoviszidose sind verschiedene Mutationen im CFTR-Gen, das beim Menschen auf dem langen Arm von Chromosom 7 (Genlocus q31.2) sitzt. Das CFTR-Gen codiert für das Protein Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Dieses Genprodukt fungiert in der Zellmembran als Chloridkanal. Durch die Veränderung im Gen wird ebenso das Protein verändert, und die Kanalfunktion bleibt aus oder ist eingeschränkt. Es handelt sich somit um eine Mutation, die zu einem Funktionsverlust des betroffenen Proteins führt (Loss-of-Function-Mutation). Die häufigste Mutation dieses Gens wird ΔF508 genannt. ΔF508 bezeichnet das Fehlen der Aminosäure Phenylalanin (‚F‘ im Einbuchstabencode) an der Position 508 im CFTR-Protein und betrifft etwa sieben von zehn Menschen mit Mukoviszidose.
Bisher sind über 2000 verschiedene Mutationen des CFTR-Gens bekannt, die in unterschiedlichen Populationen mehr oder weniger gehäuft auftreten. Eine Besonderheit dabei ist, dass bei der Mukoviszidose auch zwei verschiedene Mutationen des CFTR-Gens, also zwei verschiedene Allele desselben Gens, dennoch zur Erkrankung führen können. Diese besondere Konstellation eines autosomal-rezessiven Erbgangs wird dementsprechend Compound-Heterozygotie genannt.
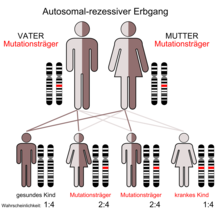
Da Mukoviszidose autosomal-rezessiv vererbt wird, tritt die Erkrankung nur dann auf, wenn der Merkmalsträger von beiden Elternteilen je ein mutiertes Gen erbt. Sind beide Elternteile Träger je eines mutierten und eines unveränderten Genes, ist die Wahrscheinlichkeit, dass ein Kind zwei intakte Genkopien erhält, 25 %. Die Wahrscheinlichkeit, dass das Kind mit einer intakten und einer mutierten Kopie zwar gesund ist, aber die Mutation weitervererben kann, ist 50 %, und die Wahrscheinlichkeit, dass das Kind erkrankt, also von beiden Eltern die krankmachende Variante erbt, beträgt ebenfalls 25 %. Sind beide Eltern erkrankt, würden auch alle Kinder die Erkrankung erben. Allerdings ist dies sehr unwahrscheinlich, da die Betroffenen meist unfruchtbar sind.
Pathologie


Das aus 1480 Aminosäuren bestehende CFTR-Protein wird von den Endothelzellen in neun bis zehn Minuten im Endoplasmatischen Retikulum synthetisiert. Die Faltung des hochkomplexen Proteins dauert etwa 30 bis 120 Minuten. Der Faltungsprozess wird durch ein ganzes Arsenal von Chaperonen, wie beispielsweise Hsp70, Hsp40, Hsp90 und Calnexin unterstützt. Liegt das Protein in diesem Zeitraum nicht korrekt gefaltet vor, so wird über Chaperone wie Hsp70 die Ubiquitin-Protein-Ligase UBR1 rekrutiert, die das falsch gefaltete CFTR ubiquitinyliert und dadurch dem Abbau im 26s-Proteasom zuführt. Diese Proteinqualitätskontrolle ist ein zellulärer Schutzmechanismus, der für die Aufrechterhaltung eines funktionierenden Proteoms und zum Überleben der Zelle von grundlegender Wichtigkeit ist. Das Kriterium, das über Abbau im Proteasom und – im Fall von CFTR – Transport zur Zellmembran entscheidet, ist allerdings nicht eine verminderte Funktion des Proteins, sondern seine signifikant reduzierte Faltungskinetik. Möglicherweise würde das Protein, wenn es ausreichend Zeit hätte, noch korrekt falten. Tatsächlich werden auch im gesunden homozygoten Menschen etwa 75 % der CFTR-Proteine von der Proteinqualitätskontrolle als „falsch“ gefaltet erkannt und abgebaut.
Das CFTR-Protein ist ein an der Zellmembran lokalisierter, durch cyclisches Adenosinmonophosphat (cAMP) regulierter Chloridkanal, der vor allem von Epithelzellen exprimiert wird. Je nach Mutation kann die Expression des Chloridkanals unterdrückt werden, oder es befinden sich nur defekte oder in ihrer Funktion oder Funktionsdauer eingeschränkte CFTR-Proteine in der Zellmembran. Der Ausfall von CFTR führt zu einer Störung des trans-epithelialen Transports in allen Organen, in denen die Epithelzellen CFTR exprimieren. Dies sind vor allem der Atemtrakt, die Bauchspeicheldrüse, der Dünndarm, die männlichen Geschlechtsorgane und die Haut. Mukoviszidose ist deshalb eine Multisystemerkrankung. Der Ausfall von CFTR hat hauptsächlich auf das Lungenepithel negative Auswirkungen. Das ist bei gesunden Menschen auf der zur Atemluft liegenden Seite mit einer etwa 5 µm starken Schicht, der Airway Surface Liquid (ASL), belegt. Diese Schicht besteht aus einem dünnflüssigen Sol und ist etwas weniger dick als die Zilienlänge (ca. 6 bis 7 µm). Auf der ASL schwimmen dickflüssige Mucine, die ein Gel bilden. Dieses Gel hat beim gesunden Menschen eine Dicke von einem bis wenigen Millimeter. Gegen inhalierte Pathogene stellt es die erste Abwehrlinie dar. Es ist ein elementarer Bestandteil der mukoziliären Clearance. Diese Abwehrlinie ist bei Patienten mit Mukoviszidose geschädigt.
Es gibt zwei unterschiedliche Hypothesen, warum der Gendefekt in CFTR zur Störung der mukoziliären Clearance führt. Beide basieren auf der Annahme, dass der primäre Defekt die Funktionsstörung von CFTR bei der Ionenaufnahme ist. Die High-Salt-Hypothese geht davon aus, dass die Epithelien durch den CFTR-Defekt nicht mehr ausreichend Natriumchlorid absorbieren. Dies führt zu einer erhöhten Konzentration von Kochsalz (high salt) in der ASL, wodurch wiederum die Wirkung von sezernierten antimikrobiellen Peptiden, wie beispielsweise β-Defensin, stark eingeschränkt ist. Nach der Low-Volume-Hypothese ist dagegen die Konzentration von Natriumchlorid in der ASL und im Plasma gleich. Allerdings soll der Ausfall von CFTR zu einer vermehrten Expression von epithelialen Natriumkanälen (ENaC) führen, was einen erhöhten Transport von Natriumionen in die Zelle zur Folge hat. Um den osmotischen Druck auszugleichen, strömen daraufhin Wassermoleküle von der basolateralen Seite in die Epithelien, was zu einer Reduktion des Volumens (low volume) der ASL führt. Die dünnere ASL bewirkt eine stark eingeschränkte mukoziliäre Clearance, wodurch die Besiedlung des Mucus mit Pathogenen erleichtert wird. Beide Hypothesen entstanden am Ende der 1990er Jahre und wurde lange Zeit kontrovers diskutiert. Aus beiden Hypothesen lassen sich unterschiedliche, sich widersprechende Behandlungsmöglichkeiten ableiten. Nach der Low-Volume-Hypothese sollte die Zufuhr von Wasser in die ASL zur Verbesserung führen, während nach der High-Salt-Hypothese die Entfernung des Kochsalzes aus der ASL die bessere Strategie wäre.
Das CFTR-Protein ist physiologisch mehr als nur ein Chloridkanal an der Zellmembran. Neben dem zuvor erwähnten Einfluss auf die Expression von epithelialen Natriumkanälen reguliert CFTR auch die Outwardly Rectifying Chloride Channels (ORCC) und den Renal Outer Medullary Potassium Channel (ROMK). Zudem wird der Chlorid-gekoppelte Hydrogencarbonat-Transport und die Aufnahme von Sphingolipiden durch CFTR beeinflusst.
Mutationsklassen

Die fast 2000 Mutationen, die Mukoviszidose verursachen, lassen sich in sechs Klassen einteilen, die sich in ihrem Pathomechanismus unterscheiden. So führen einige Mutationen zum nahezu vollständigen Ausfall der Synthese des CFTR-Proteins. Bei anderen wird beispielsweise der Einbau des Proteins in die Zellmembran verhindert, oder der Ionenkanal des Proteins ist blockiert bzw. nur eingeschränkt leitfähig. Mit Ausnahme der Gruppe der homozygoten Patienten mit ΔF508-Mutation sind die anderen Patientengruppen vergleichsweise klein. Genomweite Assoziationsstudien (GWAS), die das Ziel haben einen bestimmten Genotyp einem bestimmten Phänotyp – in diesem Fall die Ausprägung der Mukoviszidose – zuzuordnen, sind deshalb nur bei wenigen, häufigen CFTR-Mutationstypen möglich. Grundsätzlich führen die vielen unterschiedlichen Genotypen, bei denen neben den homozygoten auch noch die compound-heterozygoten CFTR-Varianten zu berücksichtigen sind, zu einer Vielzahl unterschiedlicher Phänotypen mit unterschiedlichen Krankheitsverläufen, Schweregraden und letztlich auch Lebenserwartungen.
Nicht immer ist die Klassifizierung konsistent mit dem Pathomechanismus, sondern manchmal nach dem Phänotyp gerichtet. Die Mutation A455E wird beispielsweise der Klasse V zugerechnet. Sie führt verglichen mit ΔF508 zu einer milderen Ausprägung der Mukoviszidose. Im Jahr 2014 stellte eine Arbeitsgruppe aber fest, dass bei dieser Mutation tatsächlich ein Großteil des Proteins im Proteasom abgebaut wird, was – wie im Fall von ΔF508 – einer Klasse-II-Mutation entspricht. Der Unterschied zu ΔF508 liegt offensichtlich lediglich darin, dass ein geringerer Anteil von A455E im Proteasom abgebaut wird, was letztlich zur milderen Ausprägung der Mukoviszidose bei dieser Mutation führt.
- Klasse I
Bei Klasse-I-Mutationen ist der Gendefekt so schwerwiegend, dass kein CFTR-Protein produziert wird. Die Ursache hierfür können verfrühte Stopcodons (Stoppmutationen) sein, die bei der Transkription zu einer instabilen mRNA führen. Alle Stoppmutationen, mit Ausnahme von R1162X, gehören zur Klasse I. Der nonsense-mediated mRNA Decay (NMD) ist ein zellulärer Kontrollmechanismus der vorzeitige Stopcodons in der mRNA erkennt und deren Expression als verkürzte Proteine verhindert. Beispiele für Klasse-I-Mutationen sind Gly542X, Trp1282X, und Arg553X. Eine andere Ursache können kanonische Spleißmutationen, wie zum Beispiel 621+1G→T, und chromosomale Deletionen, wie CFTRdel2,3, sein, die ebenfalls zum Ausfall der CFTR-Produktion im Endoplasmatischen Retikulum führen. Etwa 5 bis 10 % aller Mukoviszidosepatienten haben Klasse-I-Mutationen.
- Klasse II
Eine Proteinsynthese findet bei Klasse-II-Mutationen zwar statt, jedoch stimmt durch eine Mutation im CFTR-Gen die Primärstruktur des CFTR-Proteins nicht. Dies wirkt sich auf die Tertiärstruktur des Proteins aus, das Protein ist falsch gefaltet. Diese Proteinfehlfaltung wird von der Proteinqualitätskontrolle der Zelle erkannt, das Protein wird ubiquitinyliert, ins Proteasom transportiert und dort zerlegt. Die weltweit häufigste CFTR-Mutation ΔF508 gehört zu dieser Klasse, ebenso wie die Mutationen Asn1303Lys, Ile507del, Arg560Thr und Gly85Glu.
- Klasse III
Im Fall von Klasse-III-Mutationen wird das CFTR-Protein produziert und an der Zellmembran auch exprimiert. Der Chloridkanal ist allerdings nicht funktionsfähig, weil er sich nicht öffnen lässt. Beispiele für diese Klasse sind die Mutationen Gly551Asp, Gly178Arg, Gly551Ser und Ser549Asn. In Deutschland sind etwa 3 % der Mukoviszidosepatienten dieser Mutationsklasse zuzuordnen.
- Klasse IV
Auch bei Klasse-IV-Mutationen wird CFTR an der Zellmembran exprimiert. Der Chloridkanal lässt sich öffnen, allerdings ist seine Durchlässigkeit für Chloridionen stark eingeschränkt. Dies ist beispielsweise bei den Mutationen Arg117His, Arg347Pro, Arg117Cys und Arg334Trp der Fall.
- Klasse V
Bei Klasse-V-Mutationen wird zu wenig CFTR-Protein produziert. Ursache hierfür sind meist Mutationen in Introns, die zum alternativen Spleißen führen und dadurch unmittelbar negativ die Menge an produziertem CFTR-Protein beeinflussen. Beispiele hierfür sind die Mutationen 3849 + 10kbC→T, 2789 + 5G→A und 5T.
- Klasse VI
Die Mutationsklasse VI ist vergleichsweise selten. Bei ihr wird funktionsfähiges CFTR-Protein an der Zellmembran exprimiert, allerdings ist die Stabilität des Proteins an der Zellmembran reduziert, so dass es vergleichsweise schnell wieder in das Zytoplasma zurückkehrt und abgebaut wird. Ein Beispiel hierfür ist die Mutation 4326delTC. Im Vergleich zum Wildtyp hat dieses Genprodukt eine um den Faktor 5 bis 6 höhere Degradationsrate.
Klinisches Bild
Je nach Typ der Mutation sind die Symptome der Erkrankung mehr oder weniger stark ausgeprägt. Hat der Patient verschiedene Mutationen der CFTR-Gene beider Chromosomen, kommt es nur zur Ausprägung der Symptome des geringeren Defekts. Menschen mit wenig beeinträchtigenden Mutationen haben häufig nur Bauchspeicheldrüsenprobleme, bei schwerwiegenden Mutationen können alle genannten Symptome auftreten.
Atemtrakt


Bei Mukoviszidosepatienten ist der Schleim in den Bronchien deutlich zähflüssiger als bei gesunden Menschen. Dies führt zu chronischem Husten, Bronchiektasien, häufig wiederkehrenden Lungeninfekten und schweren Lungenentzündungen. Das zähe Sekret kann vom Flimmerepithel der Luftröhre und der Bronchien nur schwer abtransportiert werden. Daher stellt es ein gutes Nährmedium für Krankheitserreger wie Haemophilus influenzae und Staphylococcus aureus, vor allem in den ersten Jahren der Erkrankung, und im weiteren VerlaufPseudomonas aeruginosa, der Burkholderia-cepacia Komplex (bedeutsam: Burkholderia cepacia, B. cenocepacia und B. multivorans), Stenotrophomonas maltophilia und die „schwarze Hefe“ (Exophiala dermatitidis und Exophiala phaeomuriformis) dar. Häufig leiden Betroffene auch an Aspergillose. Zugenommen hatte zu Beginn des 21. Jahrhunderts auch der Nachweis von multiresistenten gramnegativen Stäbchen und Nichttuberkulösen Mykobakterien sowie Inquilinus limosus, Ralstonia-Arten und Pandoraea aus den Atemwegen. Eine Folge der häufigen und langwierigen Lungeninfekte kann zunehmende Lungeninsuffizienz sein, die sich durch chronischen Sauerstoffmangel und Atemnot bemerkbar macht. Durch den chronischen Sauerstoffmangel hat die Mehrzahl der betroffenen Menschen Uhrglasnägel und Trommelschlägelfinger. Die chronischen endobronchialen Infektionen führen im Endstadium zu Zysten, Trachealdivertikeln, Abszessen und Lungenfibrose, also zu einer extensiven Zerstörung der Atemwege, die auch meist die Ursache für die frühe Sterblichkeit von Mukoviszidosepatienten ist. Über 90 % der Patienten sterben an den Folgen der chronischen Infektion der unteren Atemwege.
Oxidativer Stress reduziert die Expression von CFTR in der Lunge. So konnten bei gesunden Rauchern, im Vergleich zu gesunden Nichtrauchern, Hinweise auf niedrigere Werte von CFTR in der Lunge gefunden werden. Oxidativer Stress wiederum kann durch Glutathion, ein körpereigenes Antioxidans, reduziert werden. Es hat eine wichtige Funktion zum Schutz der Lunge bei oxidativem Stress. Das Enzym Glutamatcysteinligase beeinflusst die Glutathion-Synthese in den Zellen. Ein Polymorphismus am GCLC-Gen, das für die Glutamatcysteinligase codiert, kann eine erhöhte Produktion von Glutathion in der Lunge bewirken. In einer Studie aus dem Jahr 2006 wurden 440 Mukoviszidose-Patienten auf den möglichen Einfluss des GCLC-Polymorphismus auf das Krankheitsbild untersucht. Dabei wurde ein signifikanter Zusammenhang zwischen dem funktionalen Polymorphismus von GCLC und der Schwere der Mukoviszidose bei Patienten mit einem CFTR-Genotyp der milden Form festgestellt.
Verdauungstrakt
Im Darm kommt es bei Säuglingen in 10 bis 20 % der Fälle zu einem Mekoniumileus. Dies ist ein schwerer Darmverschluss (Ileus), der durch die zähen ersten Faeces (Mekonium) hervorgerufen wird – häufig die erste Manifestation der Mukoviszidose. Bei Älteren finden sich in 20 % Obstruktionssyndrome durch zähflüssige Darmsekrete. Hierbei kann es im Einzelfall zur Komplikation kommen, dem sogenannten Mekoniumileus-Äquivalent. Es macht sich durch wiederholt auftretende Bauchschmerzen, tastbaren Darminhalt und einer Darmverlegung (Obturatio intestini) bemerkbar. Begleitet werden diese Symptome noch durch Erbrechen.
Auch die Funktion der Bauchspeicheldrüse ist gestört. Durch das fehlende Sekret entstehen chronische Durchfälle, Fettstuhl, Maldigestion, Mangelernährung und Verdauungsstörungen sowie Untergewicht. Die zunehmende Fibrosierung der Bauchspeicheldrüse führt zu einem Untergang der Langerhans-Inseln, wodurch es zum pankreatogenen Diabetes mellitus kommen kann. Man spricht in diesen Fällen von einem CF-assoziierten Diabetes (CFRD), der zum Diabetes mellitus Typ 3 gerechnet wird.
Etwa 75 % der Patienten über 19 Jahren haben eine gestörte Glucosetoleranz. Bereits bei Kindern ist das Risiko für einen Diabetes mellitus (Zuckerkrankheit) um den Faktor 10 höher als bei gleichaltrigen gesunden Kindern. Mit zunehmendem Alter steigt das Risiko weiter an. Meist wird eine frühzeitige Verabreichung von Insulin empfohlen, das mit seiner anabolen Wirkung auch zur Kräftigung des Patienten beiträgt.
Durch Störung der Leber- und Gallenwegsfunktion neigen betroffene Erwachsene zu Leberzirrhose und Gallensteinen. Bei 5,6 % der Patienten entwickelt sich eine Leberzirrhose, und bei bis zu 25 % bilden sich Gallensteine.
Skelettsystem

Die bei Mukoviszidosepatienten in den vergangenen Jahrzehnten erreichten erheblichen Verbesserungen in der Überlebensrate führen zum vermehrten Aufkommen von Spätkomplikationen, zu denen Osteoporose zählt. Etwa ein Drittel aller erwachsenen Patienten mit Mukoviszidose leidet auch an Osteoporose. Diese Patienten haben eine signifikant reduzierte Knochendichte. Dies erhöht das Risiko von Knochenbrüchen und die Ausbildung einer Kyphose („Buckel“).Rippenfrakturen sind bei erwachsenen Mukoviszidosepatienten um den Faktor 10 bis 100 häufiger als in einer gesunden Vergleichsgruppe.
Die genauen Ursachen für die reduzierte Knochendichte sind noch nicht ausreichend erforscht. Es handelt sich offensichtlich um ein sehr komplexes Zusammenspiel mehrerer Pathomechanismen. Eine wesentliche Rolle spielt dabei schlechter Ernährungszustand, die Malabsorption von Vitamin D und K, sowie ein verminderter Calcium- und Phosphatgehalt im Serum. Studien konnten zudem zeigen, dass während infektiöser Phasen die Knochenresorption (Knochenabbau) erhöht ist und Störungen in der Knochenbildung zu beobachten sind. So ist in solchen Phasen die Zahl der für den Knochenabbau verantwortlichen Osteoklasten signifikant erhöht. Neuere Forschungsergebnisse deuten außerdem darauf hin, dass die gestörte Funktion des CFTR-Proteins zu einer Funktionsstörung der für die Knochenbildung verantwortlichen Osteoblasten führt. Osteoblasten exprimieren CFTR. Das Fehlen des Chloridkanals führt bei Osteoblasten offensichtlich zu Störungen des Gleichgewichts zwischen Osteoprotegerin und Prostaglandin E2. Auch die langzeitige Anwendung von Medikamenten, insbesondere von Glucocorticoiden zur Verbesserung des respiratorischen Zustands, kann den Knochenabbau fördern. Dies ist vor allem bei Mukoviszidosepatienten mit einer Lungentransplantation der Fall. Sie erhalten zu Unterdrückung der Organabstoßung in der akuten Phase Glucocorticoide in hohen, danach in niedrigen Dosen.
Fortpflanzungsorgane
Erkrankte Männer sind in den meisten Fällen unfruchtbar.Spermien werden zwar normal gebildet, aber es fehlen beidseitig die Samenleiter. Dies wird als kongenitale bilaterale Aplasie des Vas deferens (CBAVD für engl. congenital bilateral aplasia of vas deferens) bezeichnet. CBAVD ist ein eigenständiges Krankheitsbild, bei dem 75 bis 80 % der betroffenen Männer Mutationen im CFTR-Gen tragen. Bei den Betroffenen können alle anderen Symptome einer Mukoviszidose fehlen. Solche Fälle können als milde Sonderform einer Mukoviszidose angesehen werden. Verantwortlich für die milde Ausprägung ist ein besonderes Mutationsspektrum, insbesondere des 5T-Allels. 70 bis 80 % der Männer mit CAVD haben zwei Mutationen im CFTR-Gen, wobei eine davon ein mildes Allel ist, das beispielsweise nur einen Aminosäureaustausch in einer Transmembranregion verursacht.
Bei Frauen ist durch zähflüssige Sekrete im Zervixkanal die Fruchtbarkeit vermindert. Etwa 50 % der Patientinnen sind empfängnisfähig. Bei stabiler Gesamtsituation ist für Mukoviszidose-Patientinnen das Austragen einer Schwangerschaft möglich.
Diagnostik
Mittels Pränataldiagnostik kann man bereits vor der Geburt eine mögliche Mukoviszidose nachweisen. Sie wird üblicherweise empfohlen, wenn in der Familie bereits ein erkranktes Kind vorhanden ist. Für die Geschwister eines Mukoviszidosepatienten, des sogenannten Indexpatienten, sowie für die Geschwister der heterozygoten Eltern kann sie ebenfalls angezeigt sein. Dies gilt auch für Mukoviszidosepatienten mit Kinderwunsch, beispielsweise über In-vitro-Fertilisation. Für entferntere Verwandte des Indexpatienten besteht in der Regel kein Grund zur Pränataldiagnostik. Für sie ist das Risiko eines Kindes mit Mukoviszidose kleiner als die allgemeine perinatale Sterblichkeit. Vor der Pränataldiagnostik ist eine eingehende klinisch-genetische Beratung notwendig, um die Eltern über alle Risiken des Eingriffs, der Erkrankung und die Möglichkeiten eines Schwangerschaftsabbruchs zu informieren. Vor der Pränataldiagnostik wird außerdem aus dem Blut des Indexpatienten und seiner Eltern eine CFTR-Genotypanalyse durchgeführt. Wenn der Mutationsgenotyp des Indexpatienten bekannt ist, ist die pränatale Diagnostik anhand der DNA aus fetalem Material möglich. Zur Probengewinnung sind zwei Verfahren etabliert: die Chorionzottenbiopsie und die Amniozentese (Fruchtwasseruntersuchung). Erstgenannte wird meist in der 10. bis 14. und letztgenannte in der 12. bis 16. Schwangerschaftswoche durchgeführt.
Der Schweißtest ist bei Verdacht auf Mukoviszidose bei Kleinkindern und Kindern das Mittel der Wahl. Dabei wird mit Hilfe des Arzneistoffs Pilocarpin die Schweißausscheidung stimuliert. Dazu wird ein schwacher Gleichstrom auf der Haut angelegt, der die Diffusion von Pilocarpin zu den Schweißdrüsen der Haut vermittelt. Dieses Verfahren wird Iontophorese genannt. Mit einer Kapillare wird eine Schweißprobe aus dem stimulierten Areal aufgesaugt und die Probe quantitativ auf den Gehalt von Natrium oder Chlorid analysiert. Liegt der Gehalt an Natriumchlorid in der Probe oberhalb von 80 mmol/l, so besteht ein erheblicher Verdacht auf Mukoviszidose. Zellen mit defektem CFTR-Kanal neigen dazu, vermehrt Na-Ionen zu speichern und Pilocarpin als Parasympathomimetikum steigert die Sekretion exokriner Drüsen, wie die der apokrinen Schweißdrüsen(-zellen), woraufhin eine größere Menge an Natrium und Chlorid als Schweiß abgegeben wird. Der Normalwert liegt im Bereich von 5 bis 55 mmol/l. Der Schweißtest wird üblicherweise an zwei Tagen wiederholt. Fällt er positiv aus oder sind die Ergebnisse nicht eindeutig, erfolgt im Normalfall eine DNA-Analyse (Gentest).
Eine aufwendigere Alternative zum Schweißtest ist die Bestimmung von immunreaktivem Trypsin im Blutserum. Er wird üblicherweise am fünften Lebenstag durchgeführt. Dazu wird Vollblut aus der Ferse entnommen und das Trypsin mittels Radioimmunassay bestimmt. Der Normalwert liegt unterhalb von 80 ng/ml. Die Ergebnisse sind aussagekräftiger als beim Schweißtest.
Die ersten Programme für ein Neugeborenenscreening (NGS) auf Mukoviszidose begannen 1981 in Neuseeland und Australien. In der Schweiz wird seit 2011 im Rahmen des Neugeborenenscreenings routinemäßig auch auf Mukoviszidose geprüft. Derzeit (Stand 2013) wird dies auch in den Vereinigten Staaten, England, Irland, Schottland, Frankreich, Österreich, Polen, in den Niederlanden sowie in Regionen Italiens und Spaniens so gehandhabt.
In Mecklenburg-Vorpommern wird seit 2012 bei allen Neugeborenen im Rahmen des Neugeborenenscreening auf Stoffwechselkrankheiten auch ein Test auf Mukoviszidose angeboten. 2013 wurden nur etwa 15 % der Neugeborenen in Deutschland auf Mukoviszidose hin untersucht. Deshalb wird nur bei etwa 58 % der Kinder mit Mukoviszidose die Erkrankung im ersten Lebensjahr erkannt. Das Durchschnittsalter bei der Diagnosestellung beträgt 4,8 Jahre. 7,6 % der Patienten sind bei der ersten Diagnosestellung 18 Jahre oder älter (Stand 2012). Eine frühzeitige Diagnosestellung ermöglicht eine zeitnahe Behandlung, mit der der Verlauf und die Lebensqualität der Patienten verbessert werden kann. Gegenüber der klinischen Diagnosestellung verspricht man sich auch einen langfristigen Kostenvorteil. Es gibt daher seit Jahren, speziell aus dem Bereich der Ärzteschaft, die Forderung nach einem bundesweiten Neugeborenenscreening. In den Vereinigten Staaten, Großbritannien, Frankreich und Australien wird ein genbasiertes Verfahren verwendet. Das deutsche Gendiagnostikgesetz gestattet dies nur unter strengen Auflagen. Auch deshalb wird in Deutschland der Trypsin-Test favorisiert.
Als wesentliches Kriterium für die Sinnhaftigkeit eines Neugeborenenscreenings gelten die von der Weltgesundheitsorganisation (WHO) gestellten Grundsätze, dass es „sich um eine ernsthafte Erkrankung handeln muss, deren Ätiologie und Pathogenese verstanden ist, die nach einem latenten oder frühsymptomatischen Stadium manifest wird, für die es die medizinischen und organisatorischen Möglichkeiten einer erfolgreichen Behandlung gibt und für die geeignete Test- und Untersuchungsmethoden zur Verfügung stehen“.
Ein allgemeines Heterozygoten-Screening mit einer eugenischen Zielsetzung wird von den meisten Genetikern, Pädiatern und vor allem erwachsenen Mukoviszidosepatienten strikt abgelehnt. Die Erfassung heterozygoter Merkmalsträger beim Trypsin-Test wird ebenfalls als problematisch angesehen. Von der European Community Concerted Action for Cystic Fibrosis kommt die Empfehlung, den Test auf CFTR-Heterozygotie auf Probanden mit positiver Familienanamnese zu beschränken.
2005 wurde die Erstdiagnose ‚Mukoviszidose‘ nur zu 6 % über ein Screening gefunden. Am häufigsten wurde die Diagnose nach pulmonalen (27,6 %) und gastrointestinalen, gepaart mit pulmonalen Beschwerden (21,6 %) gestellt. Rein gastrointestinale Beschwerden führten in 14,2 % und ein Mekoniumileus in 11,2 % zum entsprechenden Befund. In 6,7 % der Fälle war es ein Geschwisterkind.
Seit dem 1. September 2016 wird bundesweit ein generelles Neugeborenenscreening durchgeführt. Es umfasst das immunreaktive Trypsin, das pankreatitisassoziierte Protein und eine CFTR-Genetik.
Therapie
Dank Krankengymnastik, Inhalationen und Medikamenten, insbesondere durch ständig verbesserte Verdauungsenzyme und Antibiotika, die in den vergangenen Jahren auf den Markt gekommen sind, hat sich die Prognose der erkrankten Menschen in den letzten Jahren erheblich verbessert. Die Behandlung wirkt jedoch nicht ursächlich heilend, sondern nur symptomatisch. Die Mukoviszidose ist ein Systemdefekt, der verschiedene Organe betrifft. Da in etwa 97 % der Fälle eine ursächliche Behandlung (z. B. via Gentherapie) realistisch unmöglich ist, muss jede Störung der einzelnen Organsysteme noch gesondert therapiert werden.
Symptomatische Behandlung
Bei Kindern mit Gedeihverzögerung kann eine Therapie mit Wachstumshormonen indiziert sein. Ein besseres Wachstum mit verbessertem Körpergewicht führt auch zu weniger Krankenhausaufnahmen, weniger Antibiotika-Behandlungen und einer verbesserten Lungenfunktion. Die Supplementation mit den Vitaminen A, D, E und K sind ein wichtiger Bestandteil der symptomatischen Behandlung.
Die Lunge der Patienten wird häufig von immer wiederkehrenden Infekten heimgesucht, die das Lungengewebe dauerhaft schädigen. Insbesondere Problemkeime wie beispielsweise Pseudomonas aeruginosa, Burkholderia cepacia oder resistente Keime können schwere Lungenentzündungen verursachen. Große Bedeutung kommt daher der Bekämpfung dieser Keime zu. Die Lunge der meisten betroffenen Erwachsenen weist eine chronische Besiedelung mit dem Bakterium Pseudomonas aeruginosa auf, was häufig zur Verschlechterung der Lungensituation führt. Einige der genannten Bakterien, z. B. Pseudomonas aeruginosa, bilden zusammen mit dem zähen Schleim einen Biofilm in der Lunge der Erkrankten. Durch den zähen Schleim finden die Bakterien idealen Nährboden vor, in dem sie sich regelrecht verschanzen und für Antibiotika schwer zugänglich sind. Hier werden hochdosierte Antibiotika-Gaben meist intravenös und in dreimonatigen Abständen über eine Dauer von 14 Tagen verabreicht.
Bei der antimikrobiellen Behandlung der Mukoviszidose unterscheidet man vier Therapieprinzipien:
- prophylaktische Dauertherapie (kein Erregernachweis, keine Symptome)
- Frühtherapie oder Eradikationsbehandlung (Erregernachweis, keine Symptome)
- Exazerbationstherapie (mit oder ohne Erregernachweis, Symptome)
- Suppressionstherapie (chronischer Erregernachweise, keine oder chronische Symptome der Atemwege)
Neben Medikamenten zur Inhalation, die den zähen Schleim lösen, kommen auch Inhalationsmedikamente zur Erweiterung der Bronchien zum Einsatz, ebenso Antibiotika und Corticosteroide, die ebenfalls inhalativ appliziert werden. Gegen Pseudomonas aeruginosa wird meist Ciprofloxacin – für Kinder ab 5 Jahren – und Gentamicin verabreicht. Nach der Inhalation von Schleimlösern wird eine autogene Drainage oder modifizierte autogene Drainage angewendet. Beides sind speziell entwickelte Atemtechniken, die es dem Patienten ermöglichen, ohne fremde Hilfe das zähflüssige Sekret aus den tief gelegenen Atemwegen hochzubefördern und es dann abzuhusten.
Bei zunehmender Lungeninsuffizienz wird der Atemluft dauerhaft Sauerstoff zugemischt (Sauerstoff-Langzeittherapie). Unter dem Markennamen Pulmozyme wird rekombinante humane DNase (rhDNAse, Dornase alpha) als inhalatives Medikament zur Auflösung der im Schleim vorhandenen DNA-Filamente eingesetzt. Diese DNA-Filamente sind Überbleibsel von neutrophilen Granulozyten. Neutrophile Granulozyten sind Zellen des Immunsystems, die in die Lunge einwandern, um dort angesiedelte Bakterien zu attackieren. Danach werden die neutrophilen Granulozyten von anderen Zellen des Immunsystems entsorgt, wobei besagte DNA-Filamente der neutrophilen Granulozyten übrig bleiben. Diese DNA-Filamente tragen zusätzlich zur Zähigkeit des ohnehin schon zähen Schleims in der Lunge bei. Durch die Gabe von Dornase alpha wird die Spinnbarkeit des Schleims herabgesetzt und die mukoziliäre Clearance verbessert.
Bei Sonderproblemen wie Diabetes mellitus oder gestörter Produktion von Gallensäuren müssen auch diese Erkrankungen medikamentös behandelt werden. Bei auftretendem Darmverschluss, dem sogenannten Mekoniumileus-Äquivalent, muss sofort ärztliche Hilfe beansprucht werden.
Für die Behandlung von Osteoporose bei Mukoviszidose gibt es bisher noch keine Richtlinien. Der Schwerpunkt sollte bei vorbeugenden Maßnahmen liegen, die unter anderem eine gesunde Ernährung mit Calcium- und Vitamin-D-Substitution sowie körperliche Aktivität beinhalten. Eine vorhandene Osteoporose kann prinzipiell mittels Bisphosphonaten, Hormonersatztherapie oder Calcitonin behandelt werden. Die wenigen verfügbaren Studien über Bisphosphonatbehandlung bei Patienten mit Mukoviszidose belegen zwar eine Zunahme der Knochendichte, aber die Zahl der Knochenbrüche wird nicht signifikant gesenkt. Über die Behandlung der Osteoporose mit Raloxifen, Strontiumranelat und Teriparatid bei Patienten mit Mukoviszidose liegen noch keine Studienergebnisse vor. Die Gabe von Wachstumshormonen führt bei Kindern und Heranwachsenden in der Regel zu einer erhöhten Knochendichte.
Regelmäßige Kontrolluntersuchungen in einer speziellen Ambulanz im Krankenhaus, einer Uniklinik oder bei einem niedergelassenen Spezialisten sind wesentlicher Therapiebestandteil.
Im Frühjahr 2008 wurde am Klinikum Essen die Indikation für Amitriptylin auf Mukoviszidose erweitert. Amitriptylin hemmt die saure Sphingomyelinase indirekt und wirkt damit als FIASMA (Funktioneller Inhibitor der sauren Sphingomyelinase).
Unterstützende Maßnahmen
Dem durch die exokrine Pankreasinsuffizienz bedingten Gewichtsverlust wird durch die Gabe energiereicher, fettreicher Kost und die Verabreichung von Verdauungsenzymen (Pankreatine, Pilzenzyme) entgegengewirkt. Dem Körpergewicht von Mukoviszidose-Patienten kommt große Bedeutung zu. Je länger ein normales oder ideales Gewicht gehalten und Untergewicht verhindert werden kann, desto günstiger wirkt sich dies auf die Lungenfunktion aus. Erkrankte mit starkem Untergewicht weisen bei den Kontrolluntersuchungen in der Regel schlechtere Lungenfunktionswerte auf als solche mit normalem Körpergewicht oder mit nur minimalem Untergewicht. Von dieser Regel gibt es selbstverständlich Ausnahmen. Zu beachten ist, dass die erschwerte Atmung (z. B. durch Obstruktion der Lunge) den Energieumsatz abermals erhöht. Dieser Tatsache wird üblicherweise durch höhere Zufuhr von Nahrungsenergie Rechnung getragen.
Zur unterstützenden Therapie gehört regelmäßige sportliche Betätigung wie Laufen, Joggen, Radfahren, Tanzen o. ä. Die für den Einzelnen jeweils günstigste Sportart wird dem jeweiligen Gesundheitszustand angepasst und vom behandelnden Arzt empfohlen.
Lungentransplantation
Organtransplantationen, besonders von Lunge, Leber und Bauchspeicheldrüse, werden heute regelmäßig in Transplantations-Zentren durchgeführt und stellen für viele Menschen eine Alternative in der Behandlung der Mukoviszidose dar. Der Nutzen einer Lungentransplantation bei dieser Indikation ist jedoch umstritten.
Wenn die Einsekundenkapazität FEV1 der Lunge unter einen Wert von 30 % des Normbereiches sinkt und Bluthusten (Hämoptyse) gehäuft auftritt, liegt die Zweijahres-Überlebensrate bei nur noch etwa 50 %. In solchen Fällen kann eine Lungentransplantation angebracht sein. Allerdings beträgt die Wartezeit auf ein Spenderorgan ein bis drei Jahre, und nur jeder dritte bis sechste Patient kann ein Spenderorgan erhalten. Wegen des Mangels an Organspendern sterben deshalb die meisten Anwärter auf ein Spenderorgan. Üblicherweise wird eine Doppellungentransplantation durchgeführt. Dies ist notwendig, da die Immunsuppression, mit der die Abstoßung des Spenderorgans verhindert wird, die Infektionen des verbleibenden Lungenflügels forcieren würde. Außerdem würde das Spenderorgan infiziert werden. Die Dreijahresüberlebensrate nach einer Lungentransplantation liegt bei etwa 60 %.
Medikamentöse Behandlung der primären Krankheitsursache

Eine medikamentöse Behandlung der primären Krankheitsursache, also des Defektes, beziehungsweise der stark eingeschränkten Funktion des CFTR-Proteins, orientiert sich an den sechs Mutationsklassen. Für die Mutationsklassen I und II sind sogenannte Korrektoren in der Entwicklung, für Klasse-III-Mutationen Potentiatoren. Korrektoren sollen defektes CFTR korrigieren, und Potentiatoren sollen die Funktionalität oder die Anzahl der Chloridkanäle erhöhen. Ziel der Forschung ist, die CFTR-Funktion auf mindestens 5 % des Normalwertes anzuheben. Man geht davon aus, dass ab diesem Wert die Schwere der Symptome erheblich reduziert wird oder die wichtigsten Manifestationen der Krankheit eliminiert werden können.
Zugelassene Arzneimittel
Mit Ivacaftor (Kalydeco) von Vertex Pharmaceuticals wurde 2012 das erste Medikament zugelassen, das gegen die primäre Ursache einer Mukoviszidose gerichtet ist. Bis zu diesem Zeitpunkt konnte Mukoviszidose nur symptomatisch behandelt werden. Ivacaftor wurde im Januar 2012 von der Food and Drug Administration (FDA) und im Juli desselben Jahres von der Europäischen Arzneimittel-Agentur (EMA) für die Behandlung von Patienten im Alter über sechs Jahren mit einer G551D-Mutation freigegeben. Etwa 4 bis 5 % aller Mukoviszidose-Patienten weisen diese Mutation auf. Dies entspricht etwa 3000 Patienten weltweit. In Europa haben etwa 1500 Mukoviszidose-Patienten eine G551D-Mutation. In Deutschland weisen nur für etwa 2 % der Mukoviszidose-Patienten die notwendige Indikation für eine Behandlung mit Ivacaftor auf. Mit der Zulassung von Ivacaftor begann bei der Mukoviszidose die Ära der personalisierten Medizin. Die Behandlungskosten betragen in den Vereinigten Staaten etwa 300.000 $ pro Patient und Jahr. Ivacaftor wurde mit finanzieller Unterstützung der US-amerikanischen Patientenorganisation Cystic Fibrosis Foundation entwickelt. Vertex Pharmaceuticals erhielt insgesamt 75 Millionen $. Die Foundation bekommt dafür einen Teil der Gewinne von Vertex Pharmaceuticals. In Deutschland werden die Behandlungskosten in Höhe von über 330.000 € pro Patient und Jahr von den gesetzlichen Krankenkassen voll übernommen. Im Vergleich dazu liegen die Arzneimittelkosten der Standardtherapie bei etwa 21.000 € pro Patient und Jahr. Eine Lungentransplantation wird mit etwa 150.000 € angesetzt. Die G551D-Mutation bewirkt einen Klasse-III-Defekt im CFTR-Kanal. Ivacaftor gehört zur Gruppe der CFTR-Potentiatoren. Es ist ein CFTR-Kanalöffner, der den defekten CFTR-Kanal öffnet und so die verminderte Aktivität von CFTR erhöht. Die ersten klinischen Ergebnisse sind vielversprechend. Die Patienten nehmen rasch an Gewicht zu, und die Lungenfunktion verbessert sich innerhalb weniger Wochen signifikant.
Im Februar 2014 wurde Ivacaftor für die Behandlung von Mukoviszidosepatienten mit acht weiteren Mutationen von der FDA zugelassen. Es handelt sich dabei um die Mutationen G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P und G1349D.
Zugelassen wurden auch die Kombination von Ivacaftor und dem Korrektor Tezacaftor sowie die Kombination von Ivacaftor und dem Korrektor Lumacaftor. Lumacaftor erwies sich aber als vergleichsweise schlecht verträglich, so dass weitere Korrektoren untersucht wurden. Seit dem 21. August 2020 ist in Europa eine Dreifachkombination aus den Korrektoren Tezacaftor und Elexacaftor und dem Potentiator Ivacaftor zugelassen. Diese Dreifachkombination wird als Kaftrio gehandelt. Das Medikament ist für alle Erkrankten ab 12 Jahren und mit mindestens einer F508del-Mutation zugelassen.
Experimentelle Wirkstoffe und Behandlungskonzepte
Mit der Entdeckung des CFTR-Gens 1989 eröffneten sich völlig neue Wege in der Behandlung der Mukoviszidose. Große Hoffnungen wurden anfänglich auf die Gentherapie gesetzt. 1993 wurde bei einem Mukoviszidosepatient die erste Gentherapie durchgeführt. Es folgten über 20 weitere klinische Studien. Verschiedene virale Vektoren wurden dabei für den Transport von mRNA verwendet. Bisher waren alle klinischen Studien, trotz vielversprechender präklinischer Daten, erfolglos. Die Ursachen für das Scheitern waren vielfältig und reichen von „unzureichende Effizienz der Transfektion“ über „zu geringe Wirkdauer“ bis hin zu „erhebliche entzündliche Nebenwirkungen im Zielgewebe“.
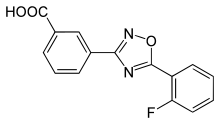
Zur Gruppe der Korrektoren in der Mutationsklasse I gehört der experimentelle Wirkstoff Ataluren. Mit Ataluren soll das korrekte Weiterlesen des CFTR-Gens über ein falsch gesetztes Stopcodon hinaus ermöglicht werden. Speziell beim Stopcodon UGA soll Ataluren besonders wirksam sein. Ataluren ist oral verfügbar, und erste klinische Studien brachten vielversprechende Ergebnisse. Derzeit (Stand März 2015) befindet sich Ataluren in der klinischen Phase III. Die Studie wird vermutlich im November 2016 beendet.
Zur Gruppe der Korrektoren gehören auch chemische Chaperone. Diese potenziellen Wirkstoffe sollen den Proteinfaltungsvorgang unterstützen beziehungsweise die richtige Proteinfaltung von CFTR ermöglichen. Sie sollen folglich Gendefekte der Klasse II kompensieren. Chemische Chaperone wirken unspezifisch auf alle zellulären Proteinfaltungsvorgänge. Eines dieser Chaperone ist Natriumphenylbutyrat. Mit diesem Wirkstoff wurden in der Vergangenheit mehrere klinische Studien durchgeführt. Dabei konnten zwar erfolgversprechende Ergebnisse erzielt werden, jedoch sind die dazu notwendigen Dosen im Bereich von 20 Gramm pro Tag extrem hoch und die Nebenwirkungen, selbst bei deutlich geringeren Dosen, beträchtlich. Dieser Therapieansatz wird deshalb nicht weiterverfolgt. Die letzte klinische Studie wurde 2011 abgebrochen. Natriumphenylbutyrat ist zur Behandlung von Patienten Hyperammonämie zugelassen. Für die Behandlung von Mukoviszidose besteht keine Zulassung. Derzeit befindet sich der potenzielle Wirkstoff Glycerin-tri-(4-phenylbutyrat), ein Triglycerid von Phenylbutyrat, in der klinischen Phase II zur Behandlung von Mukoviszidose. Diese Verbindung ist deutlich lipophiler und möglicherweise besser verträglich. Die Dosen sind ebenfalls sehr hoch und liegen im Bereich von 30 Gramm pro Tag.
Dem Furocumarin 4,6,4′-Trimethylangelicin (TMA) hat die EMA im Juni 2013 den Status eines Orphan-Arzneimittels verliehen. Von TMA wird ein bifunktionaler Wirkungsmechanismus erhofft. Die Verbindung soll zum einen die Funktion von mutiertem CFTR wiederherstellen beziehungsweise dessen Aktivität erhöhen und zum anderen entzündungshemmend wirken, indem es die Expression von Interleukin-8 herunterreguliert. TMA soll in klinischen Studien bei Patienten mit ΔF508-Mutation getestet werden.
Modellorganismen
Für die Forschung werden geeignete Tiermodelle benötigt, die der menschlichen Mukoviszidose möglichst nahekommen. Damit können die physiologischen Prozesse, die zum pathologischen Bild der Erkrankung führen, besser verstanden und vor allem neue Wirkstoffe zur Behandlung entwickelt und präklinisch getestet werden, bevor sie zur Anwendung im Menschen kommen. Das ideale Tiermodell soll die wesentlichen Merkmale wie Entzündungsprozesse in den Atemwegen, die spontane Entwicklung bakterieller Infektionen und die Progression zu einer chronischen Infektion widerspiegeln. Bereits 1992, also drei Jahre nach der Entdeckung des CFTR-Gens, wurde die Cftr-defiziente Maus entwickelt. In diesen Modellorganismus wurden große Hoffnungen gesetzt, die allerdings nicht erfüllt werden konnten. Die „Mukoviszidose-Maus“ (Cftr-Knockout) entwickelt beispielsweise keine spontanen Infekte in der Lunge. Selbst die Inokulation großer Mengen Mukoviszidose-typischer Lungenpathogene werden vom Immunsystem der Mäuse schnell beseitigt. Aus diesem Grund ist die Cftr-Knockout-Maus zur Entwicklung neuer antibakterieller oder entzündungshemmender Behandlungskonzepte wenig geeignet.
Besser geeignet sind Hausschweine. Es gibt sie seit 2008 als Cftr-defiziente Schweine und seit 2011 auch als homozygote ΔF508-Schweine. Diese Tiere entwickeln spontan die durch Infektionen, Entzündungen, starke Verschleimung und Atemwegsobstruktion gekennzeichneten, für Mukoviszidose typischen Lungenerkrankungen. Auch der Aufbau der Airway Surface Liquid von Mensch und Schwein sind einander ähnlich. Auch das seit 2010 verfügbare Mukoviszidose-Frettchen entwickelt in früher Jugend spontane Lungeninfektionen und ist als Modellorganismus in der Mukoviszidose-Forschung im Einsatz. Forschungen an beiden Tiermodellen unterstützen die Hypothese, dass das CFTR-Protein eine direkte Rolle bei der mukosalen Immunität spielt, die weit über die Rolle der Befeuchtung der Airway Surface Liquid geht.
Prognose

Während im 20. Jahrhundert noch viele Mukoviszidose-Patienten schon im Jugendalter starben, besteht heute aufgrund der sich stetig verbessernden Therapiemöglichkeiten eine gute Chance, das 40. Lebensjahr zu erreichen. Für heute Neugeborene wird bereits ein Wert von 57 Jahren angegeben. Weltweit sind mittlerweile über 50 % der Mukoviszidosepatienten über 18 Jahre alt.
Innerhalb der Mutationsklassen wird mit steigender Nummer die Prognose im Allgemeinen günstiger. Die Klassen I bis III bilden eine ‚Hochrisikogruppe‘, die Klassen IV bis VI eine ‚Niedrigrisikogruppe‘. In einer 2006 veröffentlichten Studie wurden 1672 Todesfälle von Mukoviszidosepatienten diesen beiden Risikogruppen zugeordnet. Dabei erreichten die Patienten der Niedrigrisikogruppe ein mittleres Alter von 37,6 (IQR 28,8 – 47,9) und die der Hochrisikogruppe eines von 24,2 Jahren (IQR 18,4 – 32,0). Minimal Erkrankte haben eine normale Lebenserwartung und sind in der Lage, Kinder zu zeugen oder auszutragen.
Durch die deutlich verbesserte Prognose hat sich auch das Krankheitsbild etwas gewandelt. Spätkomplikationen wie Osteoporose und Diabetes mellitus sind nun deutlich häufiger. Während Mukoviszidose früher vor allem eine Krankheit für Kinderärzte war, beschäftigt sie nun mehr und mehr Internisten und Pneumologen. Dazu kommen auch psychische Begleiterscheinungen wie Depressionen und Ängste, die bei erwachsenen Mukoviszidosepatienten weit verbreitet sind.
Heterozygote Merkmalsträger
Heterozygote CFTR-Mutationsträger können zwar nicht an einer Mukoviszidose erkranken, haben allerdings eine signifikant geringere Expression an CFTR-Protein. In einer Reihe von Studien wurde untersucht, ob dies möglicherweise anderweitige gesundheitliche Auswirkungen hat – negative wie positive.
Allgemeine Erkrankungsrisiken
Gesichert ist, dass heterozygote CFTR-Mutationsträger anfälliger für eine Pankreatitis (Bauchspeicheldrüsenentzündung) sind. Das Risiko, an einer idiopathischen chronischen Pankreatitis zu erkranken, ist etwa um den Faktor zwei bis elfmal höher als bei Menschen ohne defektes CFTR-Gen. Die genauen Ursachen hierfür sind noch unbekannt.
Bereits 1976 stellte eine Studie fest, dass heterozygote CFTR-Mutationsträger auch für Allergien anfälliger sind. Die Studienergebnisse über den Zusammenhang eines erhöhten Asthmarisikos bei heterozygoten CFTR-Mutationsträger sind dagegen bisher widersprüchlich. Sie reichen vom erhöhten Risiko bis zu leichter Schutzfunktion.
Über die Gesamtpopulation betrachtet scheint die Fortpflanzungsfähigkeit von weiblichen und männlichen Merkmalsträgern der der Restbevölkerung zu entsprechen. CFTR-Mutationen erhöhen auch nicht das Risiko, dass sich aus einer chronischen Bronchitis eine chronisch obstruktive Lungenerkrankung (COPD) entwickelt.
Merkmalsträger haben, wie auch Mukoviszidosepatienten, einen reduzierten Blutdruck. Dieser Befund wurde in der Vergangenheit über den erhöhten Elektrolytverlust erklärt. Neuere Untersuchungen aus dem Jahr 2013 zeigen jedoch, dass die reduzierte CFTR-Expression auch Veränderungen an den Blutgefäßen bewirkt. Die Agonist-induzierte Freisetzung von Calciumionen durch die glatten Muskelzellen der Aorta wird vermindert. Auch dies reduziert den Blutdruck. Der Effekt des reduzierten Blutdrucks bei Merkmalsträgern macht sich vor allem in fortgeschrittenem Alter und beim systolischen Blutdruck bemerkbar. In einer britischen Studie mit über 1200 Probandinnen lag bei heterozygoten Merkmalsträgerinnen der systolische um 7 mmHg und der diastolische Blutdruck um 4 mmHg niedriger als in der Vergleichsgruppe. Erniedrigter Blutdruck bietet erhöhten Schutz vor Schlaganfällen und koronarer Herzkrankheit. Aus den Blutdruckwerten errechneten die Autoren der Studie für heterozygote Frauen ein um 30 % reduziertes Risiko für einen Schlaganfall und ein um 20 % reduziertes Risiko für einen Myokardinfarkt.
CFTR-heterozygote Frauen haben Studien zufolge im Vergleich zu Frauen mit zwei nicht-mutierten CFTR-Allelen keine reduzierte Fruchtbarkeit.
Selektionsvorteil des heterozygoten Genotyps
Es gibt derzeit noch keine endgültige Erklärung dafür, warum ein Allel, das eine so schwerwiegende Erkrankung hervorruft, so weit verbreitet ist und nicht im Laufe der Evolution ausselektiert wurde. In kleinen, isolierten Populationen kann über Gendrift und Gründereffekt die Häufigkeit der Mukoviszidose erklärt werden. Generell kann eine tödlich verlaufende rezessive Erbkrankheit in großen Populationen über diese beiden Effekte allein nicht eine so große Häufigkeit erreichen. Alle populationsgenetischen Daten deuten auf einen Heterozygotenvorteil als Hauptursache für die hohe Frequenz der wichtigsten CFTR-Mutationen. Das heißt, dass der Funktionsverlust bei nur einem der beiden CFTR-Allele, der zur Reduzierung der Anzahl der funktionsfähigen Chlorid-Kanäle führt, einen Selektionsvorteil bewirkt. Das bekannteste Beispiel für einen Heterozygotenvorteil stellen heterozygote Merkmalsträger bei der Sichelzellenanämie dar. Sie sind weitgehend symptomlos, erkranken aber deutlich seltener an Malaria. Im Fall der Mukoviszidose wurde eine Reihe von Hypothesen aufgestellt, bei welchen Erkrankungen die heterozygote Merkmalsträger eine erhöhte Resistenz aufweisen. Diese Resistenz bietet den Selektionsvorteil, der wiederum zur ausgesprochen häufigen Verbreitung des Gendefekts geführt hat. Bis heute ist dieser Selektionsvorteil, der die Mukoviszidose zu einer der häufigsten Erbkrankheiten gemacht hat, nicht sicher bestimmt.
Vermutet werden unter anderem höhere Resistenzen gegen bestimmte Pathogene. Mit der Entdeckung, dass das Choleratoxin zu einer erhöhten CFTR-Expression in den Darmepithelien führt, was zum massiven Wasserverlust bei Cholera führt, wurde die Hypothese aufgestellt, dass hier der Selektionsvorteil für heterozygote Merkmalsträger liegt. Das Bakterium Salmonella Typhimurium gelangt über CFTR in die Epithelien, weshalb die Hypothese steht, dass die verminderte CFTR-Expression bei heterozygoten Merkmalsträgern die Wahrscheinlichkeit einer Erkrankung an Typhus reduziert. Auch dieses Pathogen fördert die Expression von CFTR. In Gebieten, in denen Typhus endemisch ist, konnte die Korrelation zwischen Erkrankungswahrscheinlichkeit und CFTR-Genotyp bestätigt werden. Allerdings ist in den untersuchten Gebieten Mukoviszidose ausgesprochen selten, und bei keinem der 775 Probanden konnte eine ΔF508-Mutation gefunden werden. Die Ausbreitungsgebiete von Cholera und Typhus korrelieren nicht mit der Häufigkeit der CFTR-Mutation in diesen Gebieten, was ein Indiz gegen diese beiden Hypothesen ist. Auch mathematische Modelle auf der Basis von historischen demographischen und epidemiologischen Daten zeigen, dass weder Cholera noch Typhus einen ausreichenden Selektionsdruck haben konnten, um die hohe Inzidenz der Mukoviszidose zu erklären.
Im Gegensatz dazu sprechen diese Modelle, zusammen mit klinischen und molekularbiologischen Daten dafür, dass bei Tuberkulose in der Vergangenheit der Selektionsdruck ausreichend hoch war. Ab dem Beginn des 16. Jahrhunderts bis zum Beginn des 20. Jahrhunderts war die Tuberkulose in Europa pandemisch („weiße Pest“) und für über 20 % der Todesfälle verantwortlich. Tuberkulose hat daher einen ausgesprochen hohen Selektionsdruck. Die Tuberkulose-Hypothese wurde bereits 1967 auf der Basis der klinischen Beobachtung aufgestellt, dass Mukoviszidosepatienten selten an Tuberkulose erkranken. Später konnte bei heterozygoten CFTR-Merkmalsträgern eine geringere Mortalität bei Tuberkulose festgestellt werden. Die Ursache für die erhöhte Resistenz von Mukoviszidosepatienten gegen Tuberkulose ist vermutlich die reduzierte Aktivität des Enzyms Arylsulfatase B. Die krankheitsauslösenden Mykobakterien haben statt Arylsulfatase B das Enzym Arylsulfotransferase, weshalb sie auf Sulfatquellen ihres Wirtes angewiesen sind, um ihre Zellwand aufzubauen. Fehlen diese Quellen, können sich die Mykobakterien nicht ausreichend vermehren. Die Tuberkulose-Hypothese erfüllt die drei Kriterien für Selektionsfaktoren: das molekularbiologische, das klinische und das historisch-geografische Kriterium. Für die Tuberkulose-Hypothese spricht auch das zum Ursprung der Mukoviszidose passende zeitnahe erstmalige Auftreten von Mycobacterium tuberculosis vor etwa 35.000 Jahren. Geht man davon aus, dass diese Tuberkulose-Hypothese korrekt ist, so müsste, mit der in entwickelten Ländern deutlich gesunkenen Tuberkulosemortalität von Menschen im zeugungsfähigen Alter, die Inzidenz für Mukoviszidose in den nächsten 100 Jahren um 0,1 % pro Jahr sinken. Um die Inzidenz zu halbieren, würde es in diesen Ländern etwa 20 Generationen benötigen.
Eine weitere Hypothese basiert auf einer Korrelation zwischen der allgemeinen adulten Milchzuckertoleranz und der Erkrankungshäufigkeit in bestimmten Völkern. So ist die Rate in der europäischen und nordamerikanischen europäischstämmigen Bevölkerung, die jeweils eine hohe Milchzuckertoleranz aufweist, am höchsten, während sie in Asien bei der dort weitverbreiteten Milchzuckerintoleranz am niedrigsten ist. Daraus ließe sich ein Zusammenhang ableiten und auch ein Selektionsvorteil für heterozygote Merkmalsträger, der den Gendefekt bisher nicht hat aussterben lassen.
Hypothesen über eine erhöhte Fortpflanzungsfähigkeit heterozygoter Merkmalsträger als Selektionsvorteil konnten epidemiologisch nicht bestätigt werden.
Medizingeschichte


Aus Mutationsfrequenzanalysen weiß man, dass die Mukoviszidose eine sehr alte Genmutation ist. Die häufigsten Mutationsarten im CFTR-Gen, wie beispielsweise ΔF508, entstanden vor ca. 51.000 Jahren im arabisch-vorderasiatischen Raum. Wahrscheinlich war die ethnische Gruppe der Belutschen die Ursprungspopulation. Die lebten zu dieser Zeit auf dem Persischen Plateau in zentraler Lage eines querenden Völkerwanderungsweges. Über diesen Weg konnte sich die Mukoviszidose durch Wanderjäger schnell nach Europa ausbreiten. Dort tauchte sie vor der letzten Eiszeit, etwa 30.000 bis 40.000 v. Chr. im Beginn des Jungpaläolithikum, erstmals auf.
Rückblickend betrachtet lässt sich das Krankheitsbild Mukoviszidose in einer Reihe von Fallbeispielen ab der Mitte des 17. Jahrhunderts in der medizinischen Literatur finden. Die Erkenntnis, dass es sich dabei um eine eigenständige Multisystemerkrankung handelt, fehlte jedoch. Die erste genauere Beschreibung der Symptome einer geschwollenen, verhärteten, weißlich schimmernden Bauchspeicheldrüse stammt aus einem Obduktionsbericht des Leidener Anatomen Peter Pauw aus dem Jahr 1595. Pauw untersuchte dabei die Leiche eines angeblich verhexten 11-jährigen Mädchens.
Schon vor Jahrhunderten wurde der salzige Geschmack von Säuglingen als unheilvolles Zeichen für die Gesundheit des Kindes und die verkürzte Lebenserwartung erkannt. Ernst Ludwig Rochholz schrieb dazu 1857 in seinem Buch Alemannisches Kinderlied und Kinderspiel aus der Schweiz:
„Das Kind stirbt bald wieder, dessen Stirne beim Küssen salzig schmeckt.“
In der Literatur wird häufig die Version aus einem Wörterbuch der Schweizerdeutschen Sprache zitiert:
„Wehe dem Kind, das beim Kuss auf die Stirn salzig schmeckt, es ist verhext und muss bald sterben.“
Der Schweizer Kinderarzt Guido Fanconi beschrieb 1936 erstmals das Krankheitsbild der Mukoviszidose als „Coeliakiesyndrom bei angeborener zystischer Pankreasfibromatose“. In der Veröffentlichung schildern Fanconi und seine beiden Co-Autoren zwei Fälle einer offensichtlich tödlichen Krankheit von Kleinkindern. Sie gingen damals davon aus, dass es sich um ein sehr seltenes Syndrom handelt.
Die Symptome der Erkrankung hatte allerdings der Österreicher Karl Landsteiner, der Entdecker der Blutgruppen, bereits 1905 beschrieben. Landsteiner schildert darin den Fall eines Mädchens, das am fünften Lebenstag „mit einem aufgetriebenen Bauch“ gestorben war. Bei der Obduktion stellte er fest, dass das Mekonium graugelb und von ausgesprochen zäher Konsistenz, wie „eingedickter Glaserkitt“, war. In diesem Zustand konnte es nicht durch die Kräfte des Darms weiterbewegt werden. Landsteiner konstatierte:
„Es ist also zu erkennen, dass die abnorme Beschaffenheit des Meconiums die letzte Ursache des Darmverschlusses bildete, da die Eindickung schon längere Zeit (also schon im Mutterleib. F. K.) bestanden hat“
Im Pankreas des Mädchens fand er eine „sehr erhebliche Vermehrung“ des Bindegewebes (Fibrose).
Den Begriff «Zystische Fibrose» (engl. cystic fibrosis) prägte die US-amerikanische Kinderärztin und Pathologin Dorothy Hansine Andersen 1938. Sie orientierte sich dabei an den Gewebeveränderungen der betroffenen Organe mit Schleimdrüsen. Außerdem definierte sie als erste das Krankheitsbild. Der US-amerikanische Pathologe Sidney Farber nannte 1944 die Erkrankung wegen der Produktion zähen Schleims «Mukoviszidose». Diese Bezeichnung hat sich vor allem im deutschsprachigen Raum durchgesetzt.
Mit der Verfügbarkeit der Antibiotika Penicillin (ab 1944), Chlortetracyclin (ab 1948), Oxytetracyclin (ab 1950), Chloramphenicol und Erythromycin (beide ab 1951) wurde die Basis der palliativen Therapie der Mukoviszidose geschaffen.
1949 erkannte Charles Upton Lowe (1921–2012), dass es sich bei der Mukoviszidose um eine Erbkrankheit handelt. Er stellte außerdem den autosomal-rezessiven Erbgang fest und postulierte, dass die Erkrankung durch einen Defekt in einem einzelnen Gen verursacht wird. Die erste Veröffentlichung über den erhöhten Elektrolytgehalt im Schweiß von Mukoviszidosepatienten stammt aus dem Jahr 1953 von Paul di Sant’Agnese (1914–2005) und Kollegen. Sie stellten bei neun Kindern mit Mukoviszidose eine um den Faktor drei erhöhte Chloridionenkonzentration im Schweiß fest. Diese Erkenntnis wird bis heute im Schweißtest zur Diagnosestellung herangezogen. Zudem wurde eine wissenschaftliche Basis für die Erkenntnisse aus dem Mittelalter über den salzigen und bitteren Geschmack des Schweißes bei Kindern mit Mukoviszidose geschaffen. Diese Erkenntnis war auch die Grundlage für die Entwicklung des Pilocarpin-Iontophorese-Schweißtestes durch Lewis E. Gibson (1927–2008) und Robert E. Cooke (1920–2014) im Jahr 1959. Die Ursache für die erhöhte Salzkonzentration fand Paul M. Quinton 1983, indem er isolierte Schweißdrüsenausführungsgänge (Ductus sudoriferus) von Mukoviszidosepatienten untersuchte und dabei eine sehr geringe Natriumchlorid-Reabsorption feststellte, die durch eine abnorm geringe Chloridionen-Permeabilität der Endothelien verursacht wird. Zwei Jahre später wurde die molekulargenetische Basis der Mukoviszidose gefunden. Eine internationale Arbeitsgruppe um Robert G. Knowlton identifizierte Chromosom 7 als Ort des Gendefekts. Dies geschah über Kopplungsanalysen bei Familien mit Kindern mit Mukoviszidose. 1989 wurde das CFTR-Gen erstmals kloniert und die Drei-Basen-Deletion ΔF508 wurde als die Mutation erkannt, die für die meisten Fälle von Mukoviszidose verantwortlich ist. Ein wichtiges Hilfsmittel bei der Suche des CFTR-Gens war die Drei-Basen-Deletion, die in etwa 70 % der damals untersuchten Mukoviszidosepatienten vorhanden war. Zum Zeitpunkt der Klonierung wusste man noch nicht, ob das CFTR-Gen für den Chloridkanal oder für einen Regulator eines Chloridkanals kodiert. Deshalb wurde der Name cystic fibrosis transmembrane conductance regulator gewählt, um beide Möglichkeiten abzudecken.
Durch die wachsenden Erkenntnisse über die Pathophysiologie der Mukoviszidose konnten in der Folgezeit neue Therapiekonzepte entwickelt werden, mit denen die Lebenserwartung der Betroffenen erheblich gesteigert werden konnte. Der vorläufige Höhepunkt ist die 2012 erfolgte Zulassung des ersten Medikaments, mit dem bei einem Teil der Patienten die Mukoviszidose ursächlich behandelt werden kann.
Für die Erforschung der zystischen Fibrose und Entwicklung von Medikamenten zur Therapie erhielten Michael J. Welsh und Paul Negulescu (Vertex Pharmaceuticals) 2022 den Shaw Prize in Life Sciences.
Siehe auch
- Lubani-al-Saleh-Teebi-Syndrom (Zystische Fibrose mit Gastritis und Megaloblastenanämie)
Rezeption
Literatur
Populärwissenschaftliche Sachbücher
- Steve Silberman: The Taste of Salt. Penguin Random House Verlagsgruppe, 2022, ISBN 978-0-593-08379-6
- Laura Rothenberg: Aufatmen (Originaltitel Breathing for a Living). Bastei Lübbe, 2003, ISBN 978-3-404-61569-8
Fachbücher
- Manfred Ballmann, Christina Smaczny, Sivagurunathan Sutharsan, Anna-Maria Dittrich (Hrsg.) CF-Manual, 3. Auflage, Thieme Verlag 2022, ISBN 978-3-13-244722-6, 240 Seiten
- Dietrich Reinhardt, Manfred Götz, Richard Kraemer, Martin H. Schöni (Hrsg.): Cystische Fibrose. Springer-Verlag, 2013, ISBN 978-3-642-56796-4, 611 S. (eingeschränkte Vorschau in der Google-Buchsuche).
- Hermann Lindemann, Burckhardt Tümmler, Gerhard Dockter (Hrsg.): Mukoviszidose – Zystische Fibrose. 4. Auflage, Georg Thieme, 2004, ISBN 3-13-138604-5, 174 S. (eingeschränkte Vorschau in der Google-Buchsuche)
- Tom O. Hirche, Thomas O. F. Wagner: Update Mukoviszidose. Georg Thieme Verlag, 2013, ISBN 978-3-13-176981-7, 136 S. (eingeschränkte Vorschau in der Google-Buchsuche)
- Margaret Hodson, Andrew Bush, Duncan Geddes: Cystic Fibrosis. 3. Auflage, CRC Press, 2012, ISBN 978-1-4441-1369-3, 486 S. (eingeschränkte Vorschau in der Google-Buchsuche)
- Deutsche Gesellschaft für Pädiatrische Infektiologie (DGPI): DGPI-Handbuch. Infektionen bei Kindern und Jugendlichen. ZDB-ID 1308754-x.
- Roland Busch: Geschichtliches über die Mukoviszidose. Hannover 1995.
Fachzeitschriften
- Journal of Cystic Fibrosis offizielles Peer-Review-Journal der European Cystic Fibrosis Society (ECFS)
Leitlinien
- S3-Leitlinie Mukoviszidose in den ersten beiden Lebensjahren (AWMF Sept. 2019)
- S3-Leitlinie Lungenerkrankung bei Mukoviszidose: Pseudomonas aeruginosa (AWMF Februar 2023)
- S1-Leitlinie Diagnose der Mukoviszidose der Deutschen Gesellschaft für Kinder- und Jugendmedizin (DGKJ). In: AWMF online (Stand 2013)
Film
- SICK: The Life and Death of Bob Flanagan, Supermasochist. 1979er Doku
- Drei Schritte zu Dir (Originaltitel: Five Feet Apart). 2019er Drama
Weblinks
- Cystic Fibrosis. In: Online Mendelian Inheritance in Man. (englisch)
- Zystische Fibrose. In: Orphanet (Datenbank für seltene Krankheiten).
- Mukoviszidose e. V. www.muko.info (Deutsche Patientenorganisation, Bundesverband seit über 50 Jahren)
- DCFH - Deutsche CF-Hilfe - Unterstützung für Menschen mit Mukoviszidose e. V.
- Cystische Fibrose – Lernprogramm für Medizinstudenten der Uni Bern
- Lungeninformationsdienst.de – Mukoviszidose
- Mukoland.de – Langzeit-Blog eines lungentransplantierten Mukoviszidose-Patienten