
| Klassifikation nach ICD-10 | |
|---|---|
| Q87.8 | Sonstige näher bezeichnete angeborene Fehlbildungssyndrome, anderenorts nicht klassifiziert |
| ICD-10 online (WHO-Version 2019) | |
Die Naxos-Krankheit, auch als Naxos-Syndrom bezeichnet, ist eine seltene autosomal-rezessiv vererbte Krankheit.
Klinisches Bild




Die Naxos-Krankheit ist äußerlich vor allem durch den kutanen Phänotyp mit wolligem Haar und Keratoderma, einer speziellen Form eines Keratoms (Keratoma hereditarium palmoplantare), an den Handflächen (palmar) und Fußsohlen (plantar) – palmoplantares Keratoderma – gekennzeichnet. Kardiologisch manifestiert sich die Naxos-Krankheit durch eine arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVCM).
Während die palmoplantare Keratoderma erst im ersten Lebensjahr auftritt, ist das wollige Haar in den meisten Fällen bereits bei der Geburt vorhanden. In der Adoleszenz zeigen alle Patienten Symptome einer Kardiomyopathie. Die Betroffenen haben Synkopen (Kreislaufkollapse) und anhaltende ventrikuläre Tachykardien („Herzrasen“). Der plötzliche Herztod ist eine häufige Folge der Erkrankung des Herzmuskels (Myokard). Die Symptome einer Rechtsherzinsuffizienz sind im Endstadium der Naxos-Krankheit zu beobachten.
Häufigkeit
Die Krankheit wurde erstmals bei Patienten von der griechischen Insel Naxos beschrieben, was der Krankheit ihren Namen gab, inzwischen sind aber auch Fälle auf anderen Inseln der Ägäis sowie in der Türkei, in Israel und in Saudi-Arabien bekannt. Die weltweite Krankheitshäufigkeit ist unbekannt. Auf Naxos wird sie auf 1:1000 geschätzt.
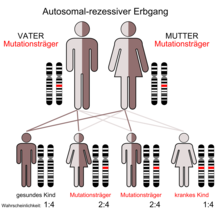
Etwa 5 % der Bevölkerung auf Naxos sind heterozygote Träger der Mutation. Abgesehen von einer kleinen Minderheit mit wolligem Haar zeigt die Mehrzahl dieser heterozygoten (mischerbigen) Menschen ein normales Erscheinungsbild. Nur wenn zwei (gesunde) Merkmalsträger ein Kind zeugen, besteht nach den Mendelschen Regeln eine statistische Wahrscheinlichkeit von 25 %, dass das Kind die Naxos-Krankheit bekommt. Zu 50 % besteht die Chance, dass es gesunder Erbträger ist und zu 25 %, dass es gesund und kein Erbträger ist.
Genetik und Pathologie
Die Ursache für die Naxos-Krankheit ist eine Mutation im JUP-Gen, das sich beim Menschen auf Chromosom 17 Genlocus q21 befindet und für das Protein Plakoglobin codiert. Plakoglobin ist ein wichtiges zytoplasmatisches Protein. Es besteht aus 744 Aminosäuren, hat eine molare Masse von 81,75 kDa und ist ein Bestandteil des Desmosoms. Als Zelladhäsionsmolekül bindet es normalerweise an das integrale Membranprotein Desmoglein I. Diese Bindung ist für den Zellkontakt wichtig. Mutationen im JUP-Gen können diesen Zellkontakt nachhaltig verändern und so die Symptome der Naxos-Krankheit auslösen.
Eine Deletion von zwei Nukleinbasen in der Position 2157–2158 im JUP-Gen konnte bei 19 Patienten mit Naxos-Krankheit festgestellt werden. Diese Deletion bewirkt einen Frameshift, der wiederum zur Folge hat, dass fünf Aminosäuren des dreizehnten Armadillo-Repeats verändert werden und die c-terminale Domäne mit 56 Aminosäuren abgeschnitten wird. Das Genprodukt faltet falsch und wird durch die Proteinqualitätskontrolle proteasomal abgebaut. Dies hat unmittelbare Auswirkungen auf die Integrität der Zellverbände. Bei der Naxos-Krankheit zeigt sich dies vor allem an der Haut und im Myokard (Herzmuskel). Werden die wegen der Mutation deutlich geschwächten zellulären Kontakte unterbrochen, so tritt der Zelltod ein und die abgestorbenen Zellen werden durch Fett und fibröses Gewebe ersetzt.
Diagnose
Die Diagnose kann anhand des auffälligen klinischen Bildes gestellt werden. Eine sichere Diagnosestellung bietet die DNA-Analyse.
Das ebenfalls autosomal-rezessiv vererbte Carvajal-Syndrom ist der Naxos-Krankheit von der Symptomatik her sehr ähnlich. Auch hier ist der Phänotyp durch Wollhaare und Keratosis palmoplantaris gekennzeichnet. Die genetische Basis des Carvajal-Syndroms sind jedoch Mutationen im DSP-Gen, das sich auf Chromosom 6 Genlocus p24 befindet und für das Zelladhäsionsprotein Desmoplakin codiert. Allerdings führt dieser Gendefekt zu einer dilatativen Kardiomyopathie, bevorzugt im linken Ventrikel, und die Kardiomyopathie tritt früher ein (early-onset).
Therapie und Prävention
Das primäre Ziel der Behandlung von Patienten mit der Naxos-Krankheit ist die Verhinderung des plötzlichen Herztods. Die Implantation eines Kardioverter-Defibrillators ist bei symptomatischen Patienten meist schon vor dem Erreichen des 35. Lebensjahrs angezeigt.
Medikamentös werden üblicherweise Antiarrhythmika, wie beispielsweise Sotalol oder Amiodaron, oft in Kombination mit klassischen Betablockern, verabreicht. Patienten mit kongestiver Herzinsuffizienz erhalten meist Diuretika und ACE-Hemmer. Im Endstadium ist eine Herztransplantation die Ultima Ratio.
Darüber hinaus gibt es Ansätze zur Prävention der Naxos-Krankheit. Durch ein systematisches Screening der Risikogruppen sollen die heterozygoten Merkmalsträger identifiziert werden.
Erstbeschreibung
Die Naxos-Krankheit wurde erstmals 1986 von einer Arbeitsgruppe um den griechischen Kardiologen Nikos Protonotarios bei einer Population auf der Insel Naxos beschrieben. Ihre Patienten mit Wollhaar und palmoplantarer Keratose zeigten kardiale Rhythmusanomalien und in den Familien traten auffällig viele Fälle von plötzlichem Herztod auf. Anhand des Stammbaums der Patienten konnten sie den autosomal-rezessiven Erbgang identifizieren. Sie untersuchten vier Familien mit insgesamt neun Fällen von Naxos-Krankheit. Der Begriff «Naxos-Krankheit» (Naxos disease) wurde 1994 durch Guy Fontaine, Nikos Protonotarios, Adalena Tsatsopoulou und Kollegen geprägt. Seit 1995 ist die Naxos-Krankheit von der Weltgesundheitsorganisation als rezessive Form der arrhythmogenen rechtsventrikulären Kardiomyopathie klassifiziert.
Weiterführende Literatur
- G. Meera, D. Prabhavathy u. a.: Naxos disease in two siblings. In: International journal of trichology. Band 2, Nummer 1, Januar 2010, S. 53–55. doi:10.4103/0974-7753.66917. PMID 21188028. PMC 3002416 (freier Volltext).
- The discovery of Naxos disease. In: European Heart Journal. Band 30, Nummer 21, November 2009, S. 2546–2548. PMID 19891084.
- N. Protonotarios, A. Tsatsopoulou: Naxos Disease. (PDF; 198 kB) In: Indian Pacing and Electrophysiology. Band 5, Nummer 2, S. 76–80.
Weblinks
- Naxos Disease. In: Online Mendelian Inheritance in Man. (englisch)
- Naxos-Krankheit. In: Orphanet (Datenbank für seltene Krankheiten).