
Als Proteinfehlfaltungserkrankungen, auch Proteinfaltungserkrankungen (engl. protein misfolding diseases oder protein misfolding disorders oder conformational diseases oder proteopathies) genannt, bezeichnet man solche Erkrankungen, die durch falsch gefaltete Proteine innerhalb und außerhalb von Zellen verursacht werden. Entweder werden die fehlgefalteten Proteine in den Zellen eingelagert oder im Proteasom abgebaut. Im ersten Fall bilden sich dabei toxische Ablagerungen (Plaques), im zweiten tritt ein Funktionsverlust, bedingt durch einen Mangel des entsprechenden Proteins in der Zelle beziehungsweise im gesamten Organismus, ein. Beides kann über die Zeit für den Betroffenen pathologisch werden und abhängig vom betroffenen Protein zu unterschiedlichen Erkrankungen führen.
Biochemischer Mechanismus
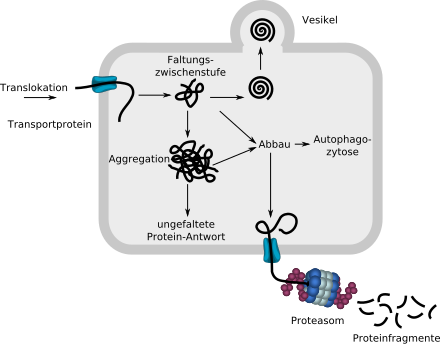
Das über ein Transportprotein in das ER eingeschleuste lineare ungefaltete Protein beginnt sich mit der Hilfe von Chaperonen (nicht eingezeichnet) zu falten. Wird es von der Proteinqualitätskontrolle als korrekt gefaltet erkannt, wird es per Vesikel aus dem ER ausgeschleust. Fehlgefaltete Proteine werden über ein Transportprotein in das Zytosol geschleust und dort in einem Proteasom in Fragmente zerlegt. Amorphe Aggregate können auch über Autophagozytose abgebaut werden.
Werden zu viele Moleküle wegen Fehlfaltung abgebaut, so kann dies zu einem Funktionsverlust in der Zelle oder dem ganzen Organismus führen. Bilden sich zu viele unlösliche, nicht mehr abbaubare Aggregate, so entstehen für die Zelle und den gesamten Organismus toxische Ablagerungen.
In den meisten Zellen aller Organismen werden im Rahmen der Proteinbiosynthese ständig die verschiedensten Proteine (Eiweiße) produziert, die in der Zelle und im gesamten Organismus die unterschiedlichsten Funktionen erfüllen. Für eine korrekte Funktion eines Proteins ist dessen Tertiärstruktur von entscheidender Bedeutung. Diese Struktur wird durch einen Prozess erreicht, der Proteinfaltung genannt wird. Die Proteinfaltung ist ein komplexer und empfindlicher Vorgang. Die korrekte Proteinfaltung wird von der Proteinqualitätskontrolle überwacht. Statistisch gesehen werden etwa 30 % aller Proteine aus der Proteinbiosynthese nicht korrekt gefaltet und normalerweise innerhalb von etwa zehn Minuten im Proteasom der Zelle abgebaut. Die Ansammlung von Proteinen mit fehlerhafter Faltung im endoplasmatischen Retikulum führt zur Unfolded Protein Response, einer Stressantwort der Zellen, die mit einer Unterdrückung der Translation und einer verstärkten Synthese von Chaperonen verbunden ist.
Ein einziges falsch gefaltetes Proteinmolekül ist nicht für die schweren Krankheitsbilder von Proteinfehlfaltungserkrankungen verantwortlich. Dafür müssen große Mengen dieser Proteine entstehen oder sich die Anzahl der korrekt gefaltete Moleküle verringern. Bei Prionen geschieht dies, weil ein falsch gefaltetes Molekül beim Kontakt mit einem korrekt gefalteten Molekül das korrekte dazu veranlasst, sich zu entfalten und zuletzt falsch wieder zusammenzufalten. Da das erste falsch gefaltete Protein durch diesen Vorgang nicht verändert wird (es funktioniert also als Enzym), sind danach zwei falsch gefaltete Moleküle vorhanden. Diese können weitere korrekte Moleküle umfalten.
Die fehlerhaften Proteine werden auch als defekte ribosomale Produkte (engl. defective ribosomal products, DRiPs) bezeichnet.
Ursachen
Die Gründe für eine falsche Proteinfaltung sind vielschichtig. Genmutationen in Exons, die zu Veränderungen in der Aminosäuresequenz, also der Primärstruktur des Genproduktes führen, haben unmittelbare Einflüsse auf die Sekundär- und Tertiärstruktur, beziehungsweise auf die Proteinfaltungskinetik. Auch Fehler bei der Transkription oder der Translation können zu Fehlfaltungen der Proteine führen. Ein weiterer möglicher Faktor ist die Umwelt; so wird bei den infektiösen Prionerkrankungen das Prion-Protein mit der Nahrung aufgenommen oder mit chirurgischem Besteck übertragen. Inzwischen gibt es auch erste Hinweise auf ein von Palmfarnen und Cyanobakterien produziertes Toxin (BMAA), das durch seinen Einbau in Proteine zu deren Fehlfaltung und so möglicherweise zu einer Form von ALS führt.
Gain-of-toxic-function
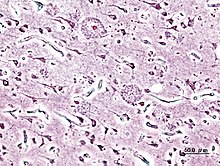
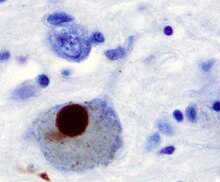
Können die DRiPs im Proteasom nicht abgebaut werden, beispielsweise, weil sie sich zuvor zu Aggregaten zusammengelagert haben, so sammeln sich die DRiPs in der Zelle an. Dort können sie mit der Zeit pathologisch werden, das heißt zu spezifischen Erkrankungen führen. Die Proteinaggregate führen vor allem zu neurodegenerativen Erkrankungen wie Parkinson-Krankheit, Alzheimer-Krankheit oder Chorea Huntington. Die Aggregate haben in den Zellen eine neue toxische Funktion. Für die toxische Wirkung innerhalb der Zellen wird der englischsprachige Begriff gain of (toxic) function verwendet.
In der englischsprachigen Fachliteratur haben sich für diese Form der Proteinfehlfaltungserkrankungen die Begriffe proteinopathy und proteopathy etabliert. Die dem entsprechenden deutschen Begriffe Proteopathie und Proteinopathie (das Präfix Proteo- = ‚Protein‘ und das Suffix -pathie = ‚Erkrankung‘) haben sich dagegen in der deutschsprachigen Fachliteratur bisher kaum durchgesetzt.
Derzeit (Stand 2011) sind über 100 Proteinopathien bei Mensch und Tier bekannt. Sie werden durch die Ablagerung von etwa 20 nicht-homologen Proteinen verursacht. Eine große und wichtige Gruppe bilden dabei die Amyloidosen.
Zu den Proteinfehlfaltungserkrankungen mit gain of toxic function zählen unter anderem folgende Erkrankungen:
Loss-of-physiological-function
Zu den Proteinfehlfaltungserkrankungen zählen außerdem die Erkrankungen, bei denen die fehlgefalteten Proteine im Proteasom zerlegt werden, wodurch keine ausreichenden Mengen des Proteins den Zellen beziehungsweise dem Organismus zur Verfügung stehen. Dieser Funktionsverlust, engl. loss of (physiological) function, kann zu Erkrankungen wie beispielsweise Mukoviszidose führen. Bei den meisten Patienten mit Mukoviszidose liegt eine ΔF508-Mutation (Typ Deletion) im CFTR-Protein – einem Chloridkanal – vor. Die Deletion von drei Nukleotiden bewirkt, dass an Position 508 von CFTR die Aminosäure Phenylalanin (im Einbuchstabencode F) fehlt. Durch diese Mutation wird das hochkomplexe CFTR, das unter anderem 21 transmembrane Proteindomänen aufweist, in seiner Faltungskinetik stark verändert. Der Faltungsprozess des CFTR-Wildtyps benötigt bereits über zwei Stunden und lediglich etwa 30 % der synthetisierten CFTR-Moleküle faltet schnell genug, um der ER-assoziierten Proteindegradation (ERAD) zu entkommen. Das ΔF508-CFTR faltet noch etwas schlechter und wird komplett abgebaut, obwohl es im Prinzip als Ionenkanal voll funktionsfähig wäre. Den von dieser Mutation betroffenen Patienten fehlt der Chloridkanal (= Funktionsverlust), was zur Folge hat, dass die Zusammensetzung der Sekrete verschiedener exkretorischer Drüsen drastisch verändert ist.
Ein Verlust an physiologischer Funktion liegt unter anderem bei folgenden Erkrankungen vor:
| Erkrankung | defektes Protein/Gen | Anmerkungen |
| Zystennieren | Polycystin-1 | |
| Morbus Charcot-Marie-Tooth | Aminoacyl-tRNA-Synthetase (AARS) | |
| X-chromosomales lymphoproliferatives Syndrom | SH2D1A | |
| Morbus Hirschsprung | Rezeptor-Tyrosinkinase Ret | |
| Homocystinurie und Methylmalonazidurie | MMACHC | |
| Patellahypoplasie | TBX4 | Ischiopatellare Dysplasie |
| Sklerosteose | Sclerostin | |
| Mukoviszidose | CFTR | |
| Phenylketonurie | Phenylalaninhydroxylase | |
| Hand-Fuß-Genital-Syndrom | Homöobox-Protein A13 | |
| lysosomale Speicherkrankheiten | verschiedene lysosomale Enzyme | über 40 einzelne Erkrankungen, u. a. Morbus Gaucher, Morbus FabryTay-Sachs-Syndrom oder Morbus Krabbe |
| QT-Syndrom | u. a. hERG | |
| Angelman-Syndrom | UBE3A | |
| erblicher Brustkrebs | BRCA1 |
Gain-of-function und Loss-of-function
Darüber hinaus gibt es Proteinfehlfaltungserkrankungen, bei denen sowohl ein Funktionsverlusts, als auch die toxischen Proteinablagerungen pathologisch werden können. Ein Beispiel hierfür ist der Alpha-1-Antitrypsin-Mangel. Eine Mutation im SERPINA1-Gen, das für das Akute-Phase-Protein α-1-Antitrypsin – ein Proteaseinhibitor – kodiert, bewirkt eine Fehlfaltung von α-1-Antitrypsin. α-1-Antitrypsin wird im Wesentlichen von Hepatozyten in der Leber exprimiert. Wegen der Fehlfaltung kann es nicht von den Heptozyten sezerniert werden und es bildet intrazelluläre Ablagerungen. Der Funktionsverlust führt bei den betroffenen Patienten zu einem progredienten Lungenemphysem, da durch den Mangel an α-1-Antitrypsin das Enzym Leukozytenelastase (engl. human leukocyte elastase, HLE) ungebremst das Lungengerüst zerstören kann. Die Ablagerungen von α-1-Antitrypsin in den Hepatozyten führen parallel zum Lungenemphysem zu einer Leberzirrhose.
Behandlungskonzepte

Die Proteinfehlfaltungerkrankungen sind derzeit nicht heilbar. Für die häufigsten neurodegenerativen Erkrankungen, die durch ein gain of toxic function verursacht werden, gibt es noch keine kausale oder kurative Therapie. Die Behandlung der Patienten erfolgt meist symptomatisch oder rein palliativ. Es gibt einige zukünftige kurative Behandlungskonzepte, wie beispielsweise die Gentherapie, die aber noch viele Jahre von einer Zulassung entfernt sind.
Proteinfehlfaltungserkrankungen, die durch einen Funktionsverlust des Proteins hervorgerufen werden, sind teilweise kurativ behandelbar. Bei der Enzymersatztherapie wird den Patienten das fehlende Protein, das gentechnisch produziert wird, mittels Infusion künstlich zugeführt. Chaperon-Therapien können für beide Arten von Proteinfehlfaltungserkrankungen zukünftige Behandlungsmöglichkeiten sein. Molekulare Chaperone sind Proteine, deren wichtigste Aufgabe es ist, neu synthetisierten Proteinen bei ihrer korrekten Faltung zu „helfen“. Darüber hinaus wurden „künstliche“ chemische und pharmakologische Chaperone identifiziert und entwickelt, die den Faltungsprozess ebenfalls unterstützen. Der Wirkstoff Sapropterin zur Behandlung der Phenylketonurie ist ein Beispiel für ein zugelassenes pharmakologisches Chaperon. Der Iminozucker 1-Deoxygalactonojirimycin (DGJ), internationaler Freiname Migalastat ist ein anderes pharmakologisches Chaperon, das derzeit (Stand Oktober 2011) in der klinischen Phase III zur Erprobung der Wirksamkeit bei Patienten mit Morbus Fabry ist.
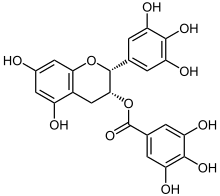
Das vor allem in grünem Tee vorkommende Epigallocatechingallat (EGCG) ist offensichtlich in der Lage, die korrekte Faltung von Proteinen zu unterstützen. Bei In-vitro-Versuchen konnte EGCG die Fibrillogenese (die Bildung von Fibrillen) von Huntingtin, α-Synuclein und β-Amyloid inhibieren. EGCG sorgt dafür, dass statt der faserförmigen toxischen Fibrillen ungefährliche sphärische Oligomere entstehen. Offensichtlich ist es auch in der Lage, bereits gebildete Plaques aufzulösen. In Farbmäusen konnte die Plaque-Belastung im Kortex, Hippocampus und im entorhinalen Kortex um jeweils etwa 50 % gesenkt werden.
Fehlfaltung außerhalb von Zellen
Seit etwa 2008 hat sich zunehmend die Erkenntnis durchgesetzt, dass Proteinfehlfaltungen nicht nur zu Problemen innerhalb von Zellen führen, sondern in bedeutendem Maße auch im Zellzwischenraum (Interstitium). Die Bedeutung des glymphatischen Systems (Abfallentsorgung des Zentralnervensystems) für den Abtransport fehlgefalteter Proteine aus dem Gehirn wurde 2012 entdeckt und ist seitdem Gegenstand intensiver Forschung. Dies betrifft insbesondere alle der bekannten und weitverbreiteten neurodegenerativen Erkrankungen.
Weiterführende Literatur
Fachbücher
- M. Ramírez-Alvarado, J. W. Kelly, C. M. Dobson (Hrsg.): Protein Misfolding Diseases. Verlag John Wiley and Sons, 2010, ISBN 0-471-79928-9 eingeschränkte Vorschau in der Google-Buchsuche
- J. Ovádi, F. Orosz (Hrsg.): Protein Folding and Misfolding: Neurodegenerative Diseases. Verlag Springer, 2009, ISBN 1-4020-9433-7 eingeschränkte Vorschau in der Google-Buchsuche
- H. J. Smith, C. Simons, R. D. E. Sewell: Protein misfolding in neurodegenerative diseases. CRC Press, 2008, ISBN 0-8493-7310-7
- V. N. Uversky, A. L. Fink (Hrsg.): Protein misfolding, aggregation and conformational diseases. Verlag Springer, 2007, ISBN 0-387-36529-X eingeschränkte Vorschau in der Google-Buchsuche
- R. M. Murphy, A. M. Tsai: Misbehaving proteins – protein (mis)folding, aggregation, and stability. Verlag Springer, 2006, ISBN 0-387-30508-4 eingeschränkte Vorschau in der Google-Buchsuche
- P. Bross, N. Gregersen (Hrsg.): Protein misfolding and disease: principles and protocols. Humana Press, 2003, ISBN 1-58829-065-4 eingeschränkte Vorschau in der Google-Buchsuche
Review-Artikel
- K. F. Winklhofer, J. Tatzelt, C. Haass: The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. In: The EMBO journal. Band 27, Nummer 2, Januar 2008, S. 336–349, ISSN 1460-2075. doi:10.1038/sj.emboj.7601930. PMID 18216876. PMC 2234348 (freier Volltext).
- H. Naiki, Y. Nagai: Molecular pathogenesis of protein misfolding diseases: pathological molecular environments versus quality control systems against misfolded proteins. In: Journal of biochemistry. Band 146, Nummer 6, Dezember 2009, S. 751–756, ISSN 1756-2651. doi:10.1093/jb/mvp119. PMID 19643812.
- L. M. Luheshi, C. M. Dobson: Bridging the gap: from protein misfolding to protein misfolding diseases. In: FEBS Letters. Band 583, Nummer 16, August 2009, S. 2581–2586, ISSN 1873-3468. doi:10.1016/j.febslet.2009.06.030. PMID 19545568.
- M. Stoppini, L. Obici u. a.: Proteomics in protein misfolding diseases. In: Clinical chemistry and laboratory medicine. Band 47, Nummer 6, 2009, S. 627–635, ISSN 1434-6621. doi:10.1515/CCLM.2009.164. PMID 19527136.
- H. Ecroyd, J. A. Carver: Unraveling the mysteries of protein folding and misfolding. In: IUBMB life. Band 60, Nummer 12, Dezember 2008, S. 769–774, ISSN 1521-6551. doi:10.1002/iub.117. PMID 18767168. (Review).
- G. B. Irvine, O. M. El-Agnaf u. a.: Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases. In: Molecular medicine (Cambridge, Mass.). Band 14, Nummer 7–8, 2008 Jul–Aug, S. 451–464, ISSN 1076-1551. doi:10.2119/2007-00100.Irvine. PMID 18368143. PMC 2274891 (freier Volltext). (Review).
- E. Laskowska, E. Matuszewska, D. Kuczynska-Wisnik: Small heat shock proteins and protein-misfolding diseases. In: Current Pharmaceutical Biotechnology. Band 11, Nummer 2, Februar 2010, S. 146–157, ISSN 1873-4316. PMID 20166966.
- N. Gregersen: Protein misfolding disorders: pathogenesis and intervention. In: Journal of inherited metabolic disease. Band 29, Nummer 2–3, 2006 Apr–Jun, S. 456–470, ISSN 1573-2665. doi:10.1007/s10545-006-0301-4. PMID 16763918. (Review).
- M. Stefani: Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. In: Biochimica et biophysica acta. Band 1739, Nummer 1, Dezember 2004, S. 5–25, ISSN 0006-3002. doi:10.1016/j.bbadis.2004.08.004. PMID 15607113. (Review).
Weblinks
- L. Walker: Proteopathies: Protein Conformational Diseases. (Memento vom 14. April 2010 im Internet Archive) Vom 18. September 2008